Pharmacology: Pharmacodynamics: Atorvastatin calcium is a synthetic lipid-lowering agent, which is an inhibitor of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase. This enzyme catalyzes the conversion of HMG-CoA to mevalonate, an early and rate-limiting step in cholesterol biosynthesis.
Mechanism of Action: Atorvastatin is a selective, competitive inhibitor of HMG-CoA reductase, the rate-limiting enzyme that converts 3-hydroxy-3-methylglutaryl-coenzyme A to mevalonate, a precursor of sterols, including cholesterol. In patients with homozygous and heterozygous familial hypercholesterolemia (FH), nonfamilial forms of hypercholesterolemia and mixed dyslipidemia, atorvastatin reduces total cholesterol (total-C), low-density lipoprotein cholesterol (LDL-C) and apolipoprotein B (apo B). Atorvastatin also reduces very-low-density lipoprotein cholesterol (VLDL-C) and triglycerides (TG) and produces variable increases in high density lipoprotein cholesterol (HDL-C).
Atorvastatin lowers plasma cholesterol and lipoprotein levels by inhibiting HMG-CoA reductase and cholesterol synthesis in the liver and by increasing the number of hepatic LDL receptors on the cell surface for enhanced uptake and catabolism of LDL.
Atorvastatin reduces LDL production and the number of LDL particles. Atorvastatin produces a profound and sustained increase in LDL receptor activity coupled with a beneficial change in the quality of circulating LDL particles. Atorvastatin is effective in reducing LDL in patients with homozygous familial hypercholesterolemia, a population that has not normally responded to lipid-lowering medication.
Atorvastatin and some of its metabolites are pharmacologically active in humans. The primary site of action of atorvastatin is the liver, which is the principal site of cholesterol synthesis and LDL clearance. Low-density lipoprotein cholesterol reduction correlates better with drug dose than it does with systemic drug concentration. Individualization of drug dosage should be based on therapeutic response (see Dosage & Administration).
In a dose-response study, atorvastatin (10-80 mg) reduced total-C (30-46%), LDL-C (41-61%), apo B (34-50%) and TG (14-33%). These results are consistent in patients with heterozygous FH, nonfamilial forms of hypercholesterolemia and mixed hyperlipidemia, including patients with non-insulin-dependent diabetes mellitus.
In patients with isolated hypertriglyceridemia, atorvastatin reduces total-C, LDL-C, VLDL-C, apo B, TG and non-HDL-C and increases HDL-C. In patients with dysbetalipoproteinemia, atorvastatin reduces IDL-C (intermediate density lipoprotein cholesterol).
In patients with Fredrickson types IIa and IIb hyperlipoproteinemia pooled from 24 controlled trials, the median percent increases from the baseline in HDL-C for atorvastatin (10-80 mg) were 5.1-8.7% in a nondose-related manner. Additionally, analysis of this pooled data demonstrated significant dose-related decreases in total-C/HDL-C and LDL-C/HDL-C ratios, ranging from -29% to -44% and -37% to -55%, respectively.
The effects of atorvastatin on ischemic events and total mortality were studied in the Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering study (MIRACL). This multicenter, randomized, double-blind, placebo-controlled study followed 3086 patients with acute coronary syndromes: Unstable angina or non-Q wave myocardial infarction. Patients were treated with standard care, including diet and either atorvastatin 80 mg daily or placebo for median duration of 16 weeks. The final LDL-C, total-C, HDL-C and TG levels were 72, 147, 48 and 139 mg/dL in the atorvastatin group, respectively, and 135, 217, 46 and 187 mg/dL, respectively, in placebo group. Atorvastatin significantly reduced the risk of ischemic events and death by 16%. The risk of experiencing rehospitalization for angina pectoris with documented evidence of myocardial ischemia was significantly reduced by 26%. Atorvastatin reduced the risk of ischemic events and death to similar extent across the range of baseline LDL-C. In addition, atorvastatin reduced the risk of ischemic events and death to similar extents in patients with non-Q wave MI and unstable angina, as well as in males and females and in patients ≤65 years and >65 years.
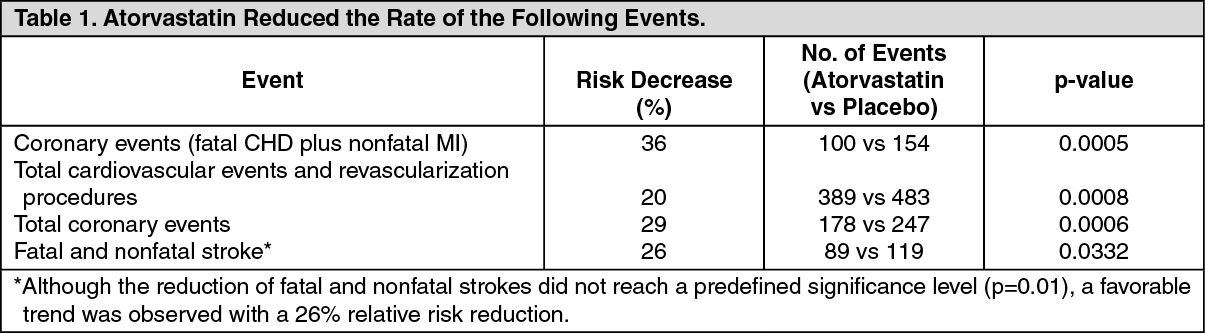
Prevention of Cardiovascular Complications: In the Anglo-Scandinavian Cardiac Outcomes Trial Lipid-Lowering Arm (ASCOT-LLA), the effect of atorvastatin on fatal and nonfatal coronary heart disease was assessed in 10,305 hypertensive patients 40-80 years (mean of 63 years), without a previous myocardial infarction and with TC levels <6.5 mmol/L (251 mg/dL). Additionally, all patients had at least 3 of the following cardiovascular risk factors: Male gender. Age >55 years, smoking, diabetes, history of chronic heart disease in a 1st degree relative, TC:HDL >6, peripheral vascular disease, left ventricular hypertrophy, prior cerebrovascular event, specific electrocardiogram abnormality, proteinuria/albuminuria. In this double-blind, placebo-controlled study patients were treated with antihypertensive therapy (goal blood pressure <140/90 mmHg for nondiabetic patients, <130/80 mmHg for diabetic patients) and allocated to either atorvastatin 10 mg daily (n=5168) or placebo (n=5137). As the effect of atorvastatin treatment compared to placebo exceeded the significance threshold during an interim analysis, the ASCOT-LLA was terminated early at 3.3 years instead of 5 years. Additionally, blood pressure was well controlled and similar in patients assigned atorvastatin and placebo. These changes persisted throughout the treatment period. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The total mortality and cardiovascular mortality have not been significantly reduced although a favorable trend was observed.
In the Collaborative Atorvastatin Diabetes Study (CARDS), the effect of atorvastatin on fatal and nonfatal cardiovascular disease was assessed in 2838 patients with type 2 diabetes 40-75 years, without prior history of cardiovascular disease and with LDL ≤4.14 mmol/L (160 mg/dL) and TG ≤6.78 mmol/L (600 mg/dL). Additionally, all patients had at least 1 of the following risk factors: Hypertension, current smoking, retinopathy, microalbuminuria or macroalbuminuria.
In this randomized, double-blind, multicenter, placebo-controlled trial, patients were treated with either atorvastatin 10 mg daily (n=1428) or placebo (n=1410) for a median follow-up of 3.9 years. As the effect of atorvastatin treatment on the primary endpoint reached the predefined stopping rules for efficacy, CARDS was terminated 2 years earlier than anticipated. (See Table 2.)
Click on icon to see table/diagram/image
There was no evidence of a difference in the treatment effect by patient’s gender, age or baseline LDL-C level.
A relative risk reduction in death of 27% (82 deaths in the placebo group compared to 61 deaths in the treatment arm) has been observed with a borderline statistical significance (p=0.0592). The overall incidence of adverse events or serious adverse events was similar between the treatment groups.
Atherosclerosis: In the Reversing Atherosclerosis with Aggressive Lipid-Lowering Study (REVERSAL), the effect of atorvastatin 80 mg and pravastatin 40 mg on coronary atherosclerosis was assessed by intravascular ultrasound (IVUS), during angiography, in patients with coronary heart disease. In this randomized, double-blind, multicenter, controlled clinical trial, IVUS was performed at baseline and at 18 months in 502 patients. In the atorvastatin group (n=253), the median percent change, from baseline, in total atheroma volume (the primary study criteria) was -0.4% (p=0.98) in the atorvastatin group and +2.7% (p=0.001) in the pravastatin group (n=249). When compared to pravastatin, the effects of atorvastatin were statistically significant (p=0.02).
In the atorvastatin group, LDL-C was reduced to a mean of 2.04 mmol/L ± 0.8 (78.9 mg/dL ± 30) from baseline 3.89 mmol/L ± 0.7 (150 mg/dL ± 28) and in the pravastatin group, LDL-C was reduced to a mean of 2.85 mmol/L ± 0.7 (110 mg/dL ± 26) from baseline 3.89 mmol/L ± 0.7 (150 mg/dL ± 26) (p<0.0001). Atorvastatin also significantly reduced mean TC by 34.1% (pravastatin: -18.4%, p<0.0001), mean TG levels by 20% (pravastatin: -6.8%, p<0.0009), and mean apolipoprotein B by 39.1% (pravastatin: -22%, p<0.0001). Atorvastatin increased mean HDL-C by 2.9% (pravastatin: +5.6%, p=NS). There was a 36.4% mean reduction in C-reactive protein (CRP) in the atorvastatin group compared to a 5.2% reduction in the pravastatin group (p<0.0001).
The safety and tolerability profiles of the 2 treatment groups were comparable.
Recurrent Stroke: In the Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) study, the effect of atorvastatin 80 mg daily or placebo on stroke was evaluated in 4731 patients who had a stroke or transient ischemic attack (TIA) within the preceding 6 months and no history of coronary heart disease (CHD). Patients were 60% male, 21-92 years (mean 63 years), and had an average baseline LDL of 133 mg/dL (3.4 mmol/L). The mean LDL-C was 73 mg/dL (1.9 mmol/L) during treatment with atorvastatin and 129 mg/dL (3.3 mmol/L) during treatment with placebo. Median follow-up was 4.9 years.
Atorvastatin 80 mg reduced the risk of the primary endpoint of fatal or nonfatal stroke by 15% (HR 0.85; 95% CI, 0.72-1; p=0.05 or 0.84; 95% CI, 0.71-0.99; p=0.03 after adjustment for baseline factors) compared to placebo. Atorvastatin 80 mg significantly reduced the risk of major coronary events (HR 0.67; 95% CI, 0.51-0.89; p=0.006), any CHD event (HR 0.6; 95% CI, 0.48-0.74; p<0.001), and revascularization procedures (HR 0.57; 95% CI, 0.44-0.74; p<0.001).
In a post-hoc analysis, atorvastatin 80 mg reduced the incidence of ischemic stroke (218/2365, 9.2% vs 274/2366, 11.6%, p=0.01) and increased the incidence of hemorrhagic stroke (55/2365, 2.3% vs 33/2366, 1.4%, p=0.02) compared to placebo. The incidence of fatal hemorrhagic stroke was similar between groups (17 atorvastatin vs 18 placebo). Reduction in the risk of cardiovascular events with atorvastatin 80 mg was demonstrated in all patient groups except in patients who entered the study with a hemorrhagic stroke and had a recurrent hemorrhagic stroke (7 atorvastatin vs 2 placebo).
In patients treated with atorvastatin 80 mg, there were fewer strokes of any type (265 atorvastatin vs 311 placebo) and fewer CHD events (123 atorvastatin vs 204 placebo). Overall mortality was similar across treatment groups (216 atorvastatin vs 211 placebo). The overall incidence of adverse events and serious adverse events was similar between treatment groups.
Secondary Prevention of Cardiovascular Events: In the Treating to New Targets Study (TNT), the effect of atorvastatin 80 mg/day versus atorvastatin 10 mg/day on the reduction in cardiovascular events was assessed in 10,001 subjects (94% white, 81% male, 38% ≥65 years) with clinically evident coronary heart disease who had achieved a target LDL-C level <130 mg/dL after completing an 8-week, open-label, run-in period with atorvastatin 10 mg/day. Subjects were randomly assigned to either 10 mg/day or 80 mg/day of atorvastatin and followed for a median duration of 4.9 years. The mean LDL-C, TC, TG, non-HDL and HDL cholesterol levels at 12 weeks were 73, 145, 128, 98 and 47 mg/dL during treatment with 80 mg of atorvastatin and 99, 177, 152, 129 and 48 mg/dL during treatment with atorvastatin 10 mg.
Treatment with atorvastatin 80 mg/day significantly reduced the rate of major cardiovascular events (MCVE) (434 events in the 80 mg/day group vs 548 events in the 10 mg/day group) with a relative risk reduction of 22%. (See Table 3.)
Click on icon to see table/diagram/image
There was no significant difference between the treatment groups for all-cause mortality: 282 (5.6%) in the atorvastatin 10 mg/day group versus 284 (5.7%) in the atorvastatin 80 mg/day group. The proportions of subjects who experienced cardiovascular death, including the components of CHD death and fatal stroke were numerically smaller in the atorvastatin 80 mg group than in the atorvastatin 10 mg treatment group. The proportions of subjects who experienced non-cardiovascular death were numerically larger in the atorvastatin 80 mg group than in the atorvastatin 10 mg treatment group.
In the Incremental Decrease in Endpoints Through Aggressive Lipid Lowering Study (IDEAL), treatment with atorvastatin 80 mg/day was compared to treatment with simvastatin 20-40 mg/day in 8888 subjects up to 80 years with a history of CHD to assess whether reduction in CV risk could be achieved. Patients were mainly male (81%), White (99%) with an average age of 61.7 years, and an average LDL-C of 121.5 mg/dL at randomization; 76% were on statin therapy. In this prospective, randomized, open-label, blinded endpoint (PROBE) trial with no run-in period, subjects were followed for a median duration of 4.8 years. The mean LDL-C, TC, TG, HDL and non-HDL cholesterol levels at week 12 were 78, 145, 115, 45 and 100 mg/dL during treatment with atorvastatin 80 mg and 105, 179, 142, 47 and 132 mg/dL during treatment with simvastatin 20-40 mg.
There was no significant difference between the treatment groups for the primary endpoint, the rate of 1st major coronary event (fatal coronary heart disease, nonfatal myocardial infarction and resuscitated cardiac arrest): 411 (9.3%) in the atorvastatin 80 mg/day group versus 463 (10.4%) in the simvastatin 20-40 mg/day group, HR 0.89, 95% CI (0.78, 1.01), p=0.07.
There were no significant differences between the treatment groups for all-cause mortality: 366 (8.2%) in the atorvastatin 80 mg/day group versus 374 (8.4%) in the simvastatin 20-40 mg/day group. The proportions of subjects who experienced CV or non-CV death were similar for the atorvastatin 80-mg group and the simvastatin 20-40 mg group.
Heterozygous Familial Hypercholesterolemia in Pediatric Patients: In a double-blind, placebo-controlled study followed by an open-label phase, 187 boys and post-menarchal girls 10-17 years (mean age of 14.1 years) with heterozygous FH or severe hypercholesterolemia were randomized to atorvastatin (n=140) or placebo (n=47) for 26 weeks and then all received atorvastatin for 26 weeks. Inclusion in the study required a baseline LDL-C level ≥190 mg/dL or a baseline LDL-C ≥160 mg/dL and positive family history of FH or documented premature cardiovascular disease in a 1st- or 2nd-degree relative. The mean baseline LDL-C value was 218.6 mg/dL (Range: 138.5-385 mg/dL) in the atorvastatin group compared to 230 mg/dL (Range: 160-324.5 mg/dL) in placebo group. The dosage of atorvastatin (once daily) was 10 mg for the first 4 weeks and up-titrated to 20 mg if the LDL-C level was >130 mg/dL. The number of atorvastatin-treated patients who required up-titration to 20 mg after week 4 during the double-blind phase was 78 (55.7%).
Atorvastatin significantly decreased plasma levels of total-C, LDL-C, triglycerides, and apolipoprotein B during the 26 week double-blind phase (see Table 4).
Click on icon to see table/diagram/image
The mean achieved LDL-C value was 130.7 mg/dL (Range: 70-242 mg/dL) in the atorvastatin group compared to 228.5 mg/dL (Range: 152-385 mg/dL) in the placebo group during the 26 week double-blind phase.
In this limited controlled study, there was no detectable effect on growth or sexual maturation in boys or on menstrual cycle length in girls. Atorvastatin has not been studied in controlled clinical trials involving pre-pubertal patients or patients <10 years. The safety and efficacy of doses >20 mg have not been studied in controlled trials in children. The long-term efficacy of atorvastatin therapy in childhood to reduce morbidity and mortality in adulthood has not been established.
Pharmacokinetics: Absorption: Atorvastatin is rapidly absorbed after oral administration; maximum plasma concentrations (C
max) occur within 1-2 hrs. Extent of absorption and plasma atorvastatin concentrations increase in proportion to atorvastatin dose. Atorvastatin tablets are 95-99% bioavailable compared with solutions. The absolute bioavailability of atorvastatin is approximately 14% and the systemic availability of HMG-CoA reductase inhibitory activity is approximately 30%. The low systemic availability is attributed to presystemic clearance in gastrointestinal mucosa and/or hepatic first-pass metabolism. Although food decreases the rate and extent of drug absorption by approximately 25% and 9%, respectively, as assessed by C
max and AUC, LDL-C reduction is similar whether atorvastatin is given with or without food. Plasma atorvastatin concentrations are lower (approximately 30% for C
max and AUC) following evening drug administration compared with morning. However, LDL-C reduction is the same regardless of the time of day of drug administration (see Dosage & Administration).
Distribution: Mean volume of distribution of atorvastatin is approximately 381 L. Atorvastatin is ≥98% bound to plasma proteins. A red blood cell/plasma ratio of approximately 0.25 indicates poor drug penetration into red blood cells.
Metabolism: Atorvastatin is extensively metabolized to ortho- and parahydroxylated derivatives and various β-oxidation products.
In vitro inhibition of HMG-CoA reductase by ortho- and parahydroxylated metabolites is equivalent to that of atorvastatin. Approximately 70% of circulating inhibitory activity for HMG-CoA reductase is attributed to active metabolites.
In vitro studies suggest the importance of atorvastatin metabolism by hepatic cytochrome P-450 3A4, consistent with increased plasma concentrations of atorvastatin in humans following co-administration with erythromycin, a known inhibitor of this isozyme.
In vitro studies also indicate that atorvastatin is a weak inhibitor of cytochrome P-450 3A4. Atorvastatin co-administration did not produce a clinically significant effect in plasma concentrations of terfenadine, a compound predominantly metabolized by cytochrome P-450 3A4; therefore, it is unlikely that atorvastatin will significantly alter the pharmacokinetics of other cytochrome P-450 3A4 substrates (see Interactions). In animals, the ortho-hydroxy metabolite undergoes further glucuronidation.
Excretion: Atorvastatin and its metabolites are eliminated primarily in bile following hepatic and/or extrahepatic metabolism; however, the drug does not appear to undergo enterohepatic recirculation. Mean plasma elimination t
½ of atorvastatin in humans is approximately 14 hrs, but the t
½ of inhibitory activity for HMG-CoA reductase is 20-30 hrs due to the contribution of active metabolites. Less than 2% of a dose of atorvastatin is recovered in urine following oral administration.
Special Populations: Elderly: Plasma concentrations of atorvastatin are higher (approximately 40% for C
max and 30% for AUC) in healthy, elderly subjects (≥65 years) than in young adults. The ACCESS study specifically evaluated elderly patients with respect to reaching their National Cholesterol Education Program (NCEP) treatment goals. The study included 1087 patients <65 years, 815 patients >65 years and 185 patients >75 years. No differences in safety, efficacy or lipid treatment goal attainment were observed between elderly patients and the overall population.
Children: Pharmacokinetic studies have not been conducted in the pediatric population.
Gender: Plasma concentrations of atorvastatin in women differ (approximately 20% higher for C
max and 10% lower for AUC) from those in men. However, there were no clinically significant differences in lipid effects between men and women.
Renal Insufficiency: Renal disease has no influence on the plasma concentrations or lipid effects of atorvastatin. Thus, dose adjustment in patients with renal dysfunction is not necessary (see Dosage & Administration).
Hemodialysis: While studies have not been conducted in patients with end-stage renal disease, hemodialysis is not expected to significantly enhance clearance of atorvastatin since the drug is extensively bound to plasma proteins.
Hepatic Insufficiency: Plasma concentrations of atorvastatin are markedly increased (approximately 16-fold in C
max and 11-fold in AUC) in patients with chronic alcoholic liver disease (Child-Pugh B) (see Contraindications).
Drug Interactions: The effect of co-administered drugs on the pharmacokinetics of atorvastatin as well as the effect of atorvastatin on the pharmacokinetics of co-administered drugs are summarized as follows (see Precautions and Interactions). (See Tables 5 and 6.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Toxicology: Preclinical Safety Data: Carcinogenesis, Mutagenesis & Impairment of Fertility: Atorvastatin was not carcinogenic in rats. The maximum dose used was 63-fold higher than the highest human dose (80 mg/day) on a mg/kg body weight basis and 8 to 16-fold higher based on AUC
(0-24) values. In a 2-year study in mice, incidences of hepatocellular adenomas in males and hepatocellular carcinomas in females were increased at the maximum dose used, which was 250-fold higher than the highest human dose on a mg/kg body weight basis. Systemic exposure was 6- to 11-fold higher based on AUC
(0-24).
All other chemically similar drugs in this class have induced tumors in both mice and rats at multiples of 12-125 times their highest recommended clinical doses, on a mg/kg body weight basis.
Atorvastatin did not demonstrate mutagenic or clastogenic potential in 4
in vitro tests with and without metabolic activation or in 1
in vivo assay. It was negative in the Ames test with
Salmonella typhimurium and
Escherichia coli, and in the
in vitro HGPRT forward mutation assay in Chinese hamster lung cells. Atorvastatin did not produce significant increases in chromosomal aberrations in the
in vitro Chinese hamster lung cell assay and was negative in the
in vivo mouse micronucleus test.
No adverse effects on fertility or reproduction were observed in male rats given doses of atorvastatin up to 175 mg/kg/day or in female rats given doses up to 225 mg/kg/day. These doses are 100-140 times the maximum recommended human dose on a mg/kg basis. Atorvastatin caused no adverse effects on sperm or semen parameters, or on reproductive organ histopathology in dogs given doses of 10, 40 or 120 mg/kg for 2 years.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image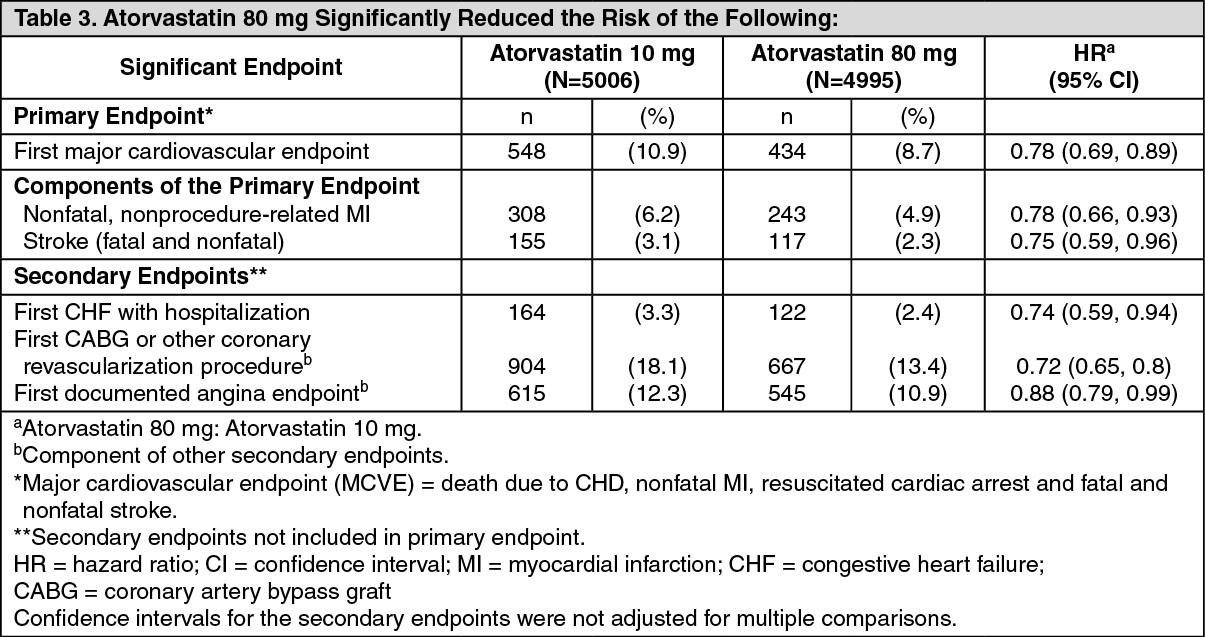 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image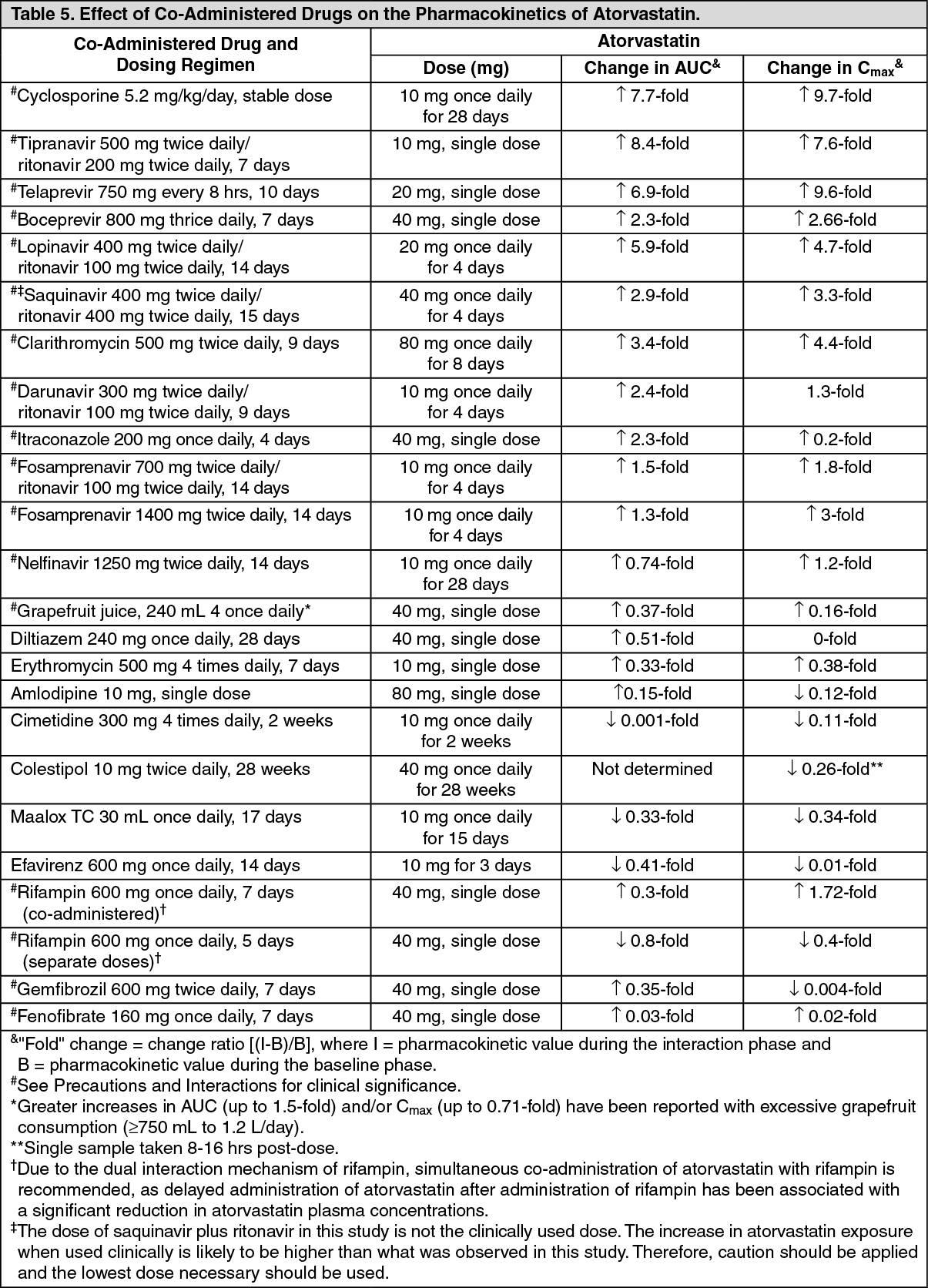 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image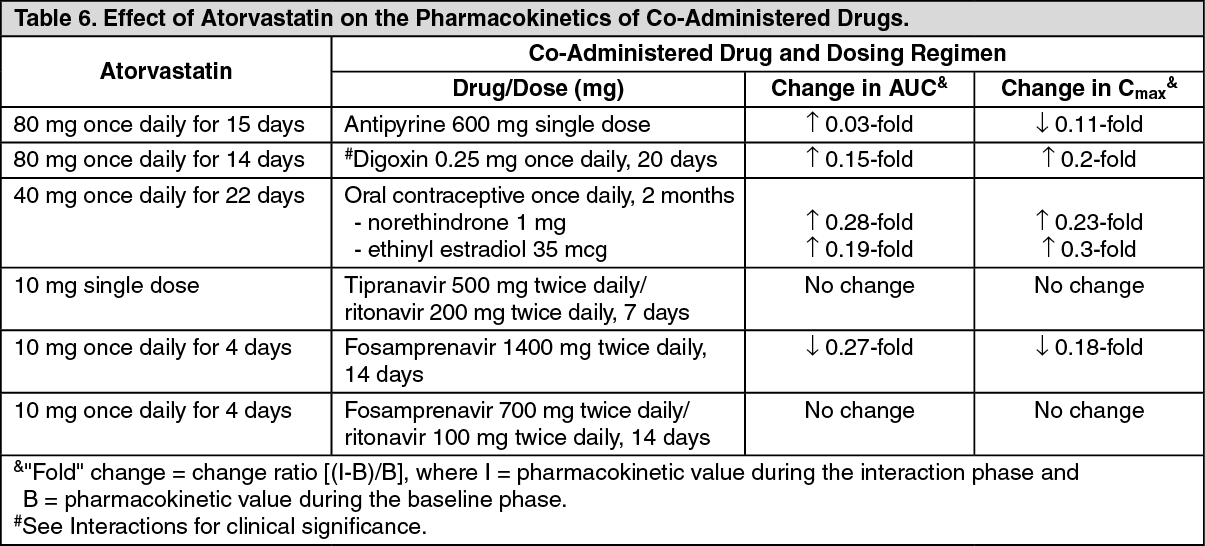 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image



 Sign Out
Sign Out
