Pharmacotherapeutic group: Antineoplastic agents, antimetabolites.
ATC code: L01BC59.
Pharmacology: Pharmacodynamics: Mechanism of action: Lonsurf is comprised of an antineoplastic thymidine-based nucleoside analogue, trifluridine, and the thymidine phosphorylase (TPase) inhibitor, tipiracil hydrochloride, at a molar ratio 1:0.5 (weight ratio, 1:0.471).
Following uptake into cancer cells, trifluridine, is phosphorylated by thymidine kinase, further metabolised in cells to a deoxyribonucleic acid DNA substrate, and incorporated directly into DNA, thereby interfering with DNA function to prevent cell proliferation.
However, trifluridine is rapidly degraded by TPase and readily metabolised by a first-pass effect following oral administration, hence the inclusion of the TPase inhibitor, tipiracil hydrochloride.
In nonclinical studies, trifluridine/tipiracil hydrochloride demonstrated antitumour activity against both 5-fluorouracil (5-FU) sensitive and resistant colorectal cancer cell lines.
The cytotoxic activity of trifluridine/tipiracil hydrochloride against several human tumour xenografts correlated highly with the amount of trifluridine incorporated into DNA, suggesting this as the primary mechanism of action.
Pharmacodynamic effects: Lonsurf had no clinically relevant effect on QT/QTc prolongation compared with placebo in an open label study in patients with advanced solid tumours.
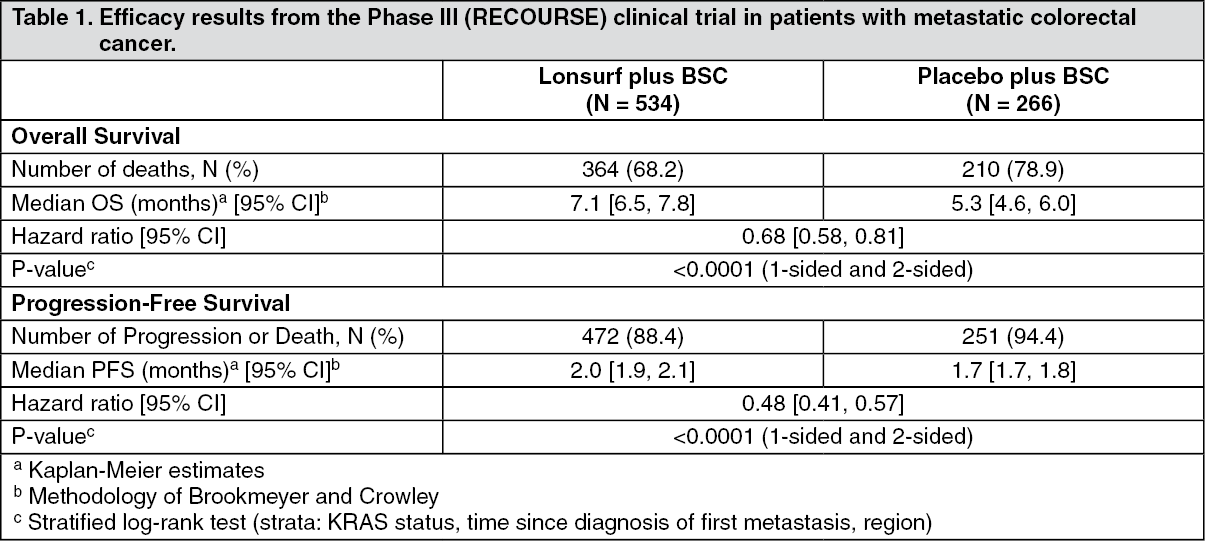
Clinical efficacy and safety: Metastatic colorectal cancer: The clinical efficacy and safety of Lonsurf were evaluated in an international, randomised, double-blind, placebo-controlled Phase III study (RECOURSE) in patients with previously treated metastatic colorectal cancer. The primary efficacy endpoint was overall survival (OS), and supportive efficacy endpoints were progression-free survival (PFS), overall response rate (ORR) and disease control rate (DCR).
In total, 800 patients were randomised 2:1 to receive Lonsurf (N = 534) plus best supportive care (BSC) or matching placebo (N = 266) plus BSC. Lonsurf dosing was based on BSA with a starting dose of 35 mg/m
2/dose. Study treatment was administered orally twice daily after morning and evening meals for 5 days a week with 2 days rest for 2 weeks, followed by 14 days rest, repeated every 4 weeks. Patients continued therapy until disease progression or unacceptable toxicity (see Dosage & Administration).
Of the 800 randomised patients, the median age was 63 years, 61% were male, 58% were Caucasian/White, 35% were Asian/Oriental, and 1% were Black/African American, and all patients had baseline Eastern Cooperative Oncology Group (ECOG) Performance Status (PS) of 0 or 1. The primary site of disease was colon (62%) or rectum (38%). KRAS status was wild (49%) or mutant (51%) at study entry. The median number of prior lines of therapy for metastatic disease was 3. All patients received prior treatment with fluoropyrimidine-, oxaliplatin-, and irinotecan-based chemotherapy. All but 1 patient received bevacizumab, and all but 2 patients with KRAS wild type tumours received panitumumab or cetuximab. The 2 treatment groups were comparable with respect to demographic and baseline disease characteristics.
An OS analysis of the study, carried out as planned at 72% (N = 574) of events, demonstrated a clinically meaningful and statistically significant survival benefit of Lonsurf plus BSC compared to placebo plus BSC (hazard ratio: 0.68; 95% confidence interval [CI] [0.58 to 0.81]; p < 0.0001) and a median OS of 7.1 months vs 5.3 months, respectively; with 1-year survival rates of 26.6% and 17.6%, respectively. PFS was significantly improved in patients receiving Lonsurf plus BSC (hazard ratio: 0.48; 95% CI [0.41 to 0.57]; p < 0.0001 (see Table 1, Figures 1 and 2).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
An updated OS analysis, carried out at 89% (N = 712) of events, confirmed the clinically meaningful and statistically significant survival benefit of Lonsurf plus BSC compared to placebo plus BSC (hazard ratio: 0.69; 95% CI [0.59 to 0.81]; p < 0.0001) and a median OS of 7.2 months vs 5.2 months; with 1-year survival rates of 27.1% and 16.6%, respectively.
The OS and PFS benefit was observed consistently, in all relevant pre-specified subgroups, including race, geographic region, age (< 65; ≥ 65), sex, ECOG PS, KRAS status, time since diagnosis of first metastasis, number of metastatic sites, and primary tumour site. The Lonsurf survival benefit was maintained after adjusting for all significant prognostic factors, namely, time since diagnosis of first metastasis, ECOG PS and number of metastatic sites (hazard ratio: 0.69; 95% CI [0.58 to 0.81]).
Sixty one percent (61%, N = 485) of all randomised patients received a fluoropyrimidine as part of their last treatment regimen prior to randomisation, of which 455 (94%) were refractory to the fluoropyrimidine at that time. Among these patients, the OS benefit with Lonsurf was maintained (hazard ratio: 0.75, 95% CI [0.59 to 0.94]).
Eighteen percent (18%, N = 144) of all randomised patients received regorafenib prior to randomisation. Among these patients, the OS benefit with Lonsurf was maintained (hazard ratio: 0.69, 95% CI [0.45 to 1.05]). The effect was also maintained in regorafenib-naive patients (hazard ratio: 0.69, 95% CI [0.57 to 0.83]).
The DCR (complete response or partial response or stable disease) was significantly higher in patients treated with Lonsurf (44% vs 16%, p < 0.0001).
Treatment with Lonsurf plus BSC resulted in a statistically significant prolongation of PS < 2 in comparison to placebo plus BSC. The median time to PS ≥ 2 for the Lonsurf group and placebo group was 5.7 months and 4.0 months, respectively, with a hazard ratio of 0.66 (95% CI: [0.56, 0.78]), p < 0.0001.
Metastatic gastric cancer: The clinical efficacy and safety of Lonsurf were evaluated in an international, randomised, double-blind, placebo-controlled Phase III study (TAGS) in patients with previously treated metastatic gastric cancer (including adenocarcinoma of the gastroesophageal junction), who had been previously treated with at least two prior systemic treatment regimens for advanced disease, including fluoropyrimidine-, platinum-, and either taxane- or irinotecan-based chemotherapy, plus if appropriate human epidermal growth factor receptor 2 (HER2)-targeted therapy. The primary efficacy endpoint was overall survival (OS), and supportive efficacy endpoints were progression-free survival (PFS), overall response rate (ORR), disease control rate (DCR), time to deterioration of ECOG performance status ≥2 and Quality of Life (QoL). Tumor assessments, according to the Response Evaluation Criteria in Solid Tumours (RECIST), version 1.1, were performed by the investigator/local radiologist every 8 weeks.
In total, 507 patients were randomised 2:1 to receive Lonsurf (N = 337) plus best supportive care (BSC) or placebo (N = 170) plus BSC. Lonsurf dosing was based on BSA with a starting dose of 35 mg/m
2/dose. Study treatment was administered orally twice daily after morning and evening meals for 5 days a week with 2 days rest for 2 weeks, followed by 14 days rest, repeated every 4 weeks. Patients continued therapy until disease progression or unacceptable toxicity (see Dosage & Administration).
Of the 507 randomised patients, the median age was 63 years, 73% were male, 70% were White, 16% were Asian, and <1% were Black/African American, and all patients had baseline Eastern Cooperative Oncology Group (ECOG) Performance Status (PS) of 0 or 1. Primary cancer was gastric (71.0%) or gastroesophageal junction cancer (28.6%) or both (0.4%). The median number of prior regimens of therapy for metastatic disease was 3. Nearly all (99.8%) patients received prior fluoropyrimidine, 100% received prior platinum therapy and 90.5% received prior taxane therapy. Approximately half (55.4%) of patients received prior irinotecan, 33.3% received prior ramucirumab, and 16.6% received prior HER2-targeted therapy. The 2 treatment groups were comparable with respect to demographic and baseline disease characteristics.
An OS analysis of the study, carried out as planned at 76% (N = 384) of events, demonstrated that Lonsurf plus BSC resulted in a statistically significant improvement in OS compared to placebo plus BSC with an hazard ratio (HR) of 0.69 (95% CI: 0.56, 0.85; 1- and 2-sided p-values were 0.0003 and 0.0006, respectively) corresponding to a 31% reduction in the risk of death in the Lonsurf group. The median OS was 5.7 months (95% CI: 4.8, 6.2) for the Lonsurf group versus 3.6 months (95% CI: 3.1, 4.1) for the placebo group; with 1-year survival rates of 21.2% and 13.0%, respectively.
PFS was significantly improved in patients receiving Lonsurf plus BSC compared to placebo plus BSC (HR of 0.57; 95% CI [0.47 to 0.70]; p < 0.0001 (see Table 2, Figures 3 and 4).
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The OS and PFS benefit was observed consistently, in all randomization strata and across most pre-specified subgroups, including sex, age (< 65; ≥ 65 years), ethnic origin, ECOG PS, prior ramucirumab treatment, prior irinotecan treatment, number of prior regimens (2; 3; ≥ 4), previous gastrectomy, primary tumour site (gastric; gastroesophageal junction) and HER2 status.
The ORR (complete response + partial response) was not significantly higher in patients treated with Lonsurf (4.5% vs 2.1%, p-value = 0.2833) but the DCR (complete response or partial response or stable disease) was significantly higher in patients treated with Lonsurf (44.1% vs 14.5%, p < 0.0001).
The median time to deterioration of ECOG performance status to ≥ 2 was 4.3 months for the Lonsurf group versus 2.3 months for the placebo group with HR of 0.69 (95% CI: 0.562, 0.854), p-value = 0.0005.
Elderly: There is limited data in Lonsurf treated patients aged 75 years and above (87 patients (10%) in pooled data of the RECOURSE and TAGS studies, of which 2 patients were 85 years or older). The effect of Lonsurf on overall survival was similar in patients < 65 years and ≥ 65 years of age.
Pharmacokinetics: Absorption: After oral administration of Lonsurf with [
14C]-trifluridine, at least 57% of the administered trifluridine was absorbed and only 3% of the dose was excreted into faeces. After oral administration of Lonsurf with [
14C]-tipiracil hydrochloride, at least 27% of the administered tipiracil hydrochloride was absorbed and 50% of the total radioactivity dose measured into faeces, suggestive of moderate gastrointestinal absorption of tipiracil hydrochloride.
Following a single dose of Lonsurf (35 mg/m
2) in patients with advanced solid tumours, the mean times to peak plasma concentrations (t
max) of trifluridine and tipiracil hydrochloride were around 2 hours and 3 hours, respectively.
In the pharmacokinetic (PK) analyses of the multiple dose administration of Lonsurf (35 mg/m
2/dose, twice daily for 5 days a week with 2 days rest for 2 weeks followed by a 14-day rest, repeated every 4 weeks), trifluridine area under the concentration-time curve from time 0 to the last measurable concentration (AUC
0-last) was approximately 3-fold higher and maximum concentration (C
max) was approximately 2-fold higher after multiple dose administration (Day 12 of Cycle 1) of Lonsurf than after single-dose (Day 1 of Cycle 1).
However, there was no accumulation for tipiracil hydrochloride, and no further accumulation of trifluridine with successive cycles (Day 12 of Cycles 2 and 3) of administration of Lonsurf. Following multiple doses of Lonsurf (35 mg/m
2/dose twice daily) in patients with advanced solid tumours, the mean times to peak plasma concentrations (t
max) of trifluridine and tipiracil hydrochloride were around 2 hours and 3 hours, respectively.
Contribution of tipiracil hydrochloride: Single-dose administration of Lonsurf (35 mg/m
2/dose) increased the mean AUC
0-last of trifluridine by 37-fold and C
max by 22-fold with reduced variability compared to trifluridine alone (35 mg/m
2/dose).
Effect of food:
When Lonsurf at a single dose of 35 mg/m
2 was administered to 14 patients with solid tumours after a standardised high-fat, high-calorie meal, trifluridine area under the concentration-time curve (AUC) did not change, but trifluridine C
max, tipiracil hydrochloride C
max and AUC decreased by approximately 40% compared to those in a fasting state. In clinical studies Lonsurf was administered within 1 hour after completion of the morning and evening meals (see Dosage & Administration).
Distribution: The protein binding of trifluridine in human plasma was over 96% and trifluridine bound mainly to human serum albumin. Plasma protein binding of tipiracil hydrochloride was below 8%. Following a single dose of Lonsurf (35 mg/m
2) in patients with advanced solid tumours, the apparent volume of distribution (Vd/F) for trifluridine and tipiracil hydrochloride was 21 L and 333 L, respectively.
Biotransformation: Trifluridine was mainly eliminated by metabolism via TPase to form an inactive metabolite, FTY. The absorbed trifluridine was metabolised, and excreted into urine as FTY and trifluridine glucuronide isomers. Other minor metabolites, 5-carboxyuracil and 5-carboxy-2'-deoxyuridine, were detected, but those levels in plasma and urine were at low or trace levels.
Tipiracil hydrochloride was not metabolised in human liver S9 or in cryopreserved human hepatocytes. Tipiracil hydrochloride was the major component and 6-hydroxymethyluracil was the major metabolite consistently in human plasma, urine, and faeces.
Elimination: Following the multiple-dose administration of Lonsurf at the recommended dose and regimen, the mean elimination half-life (t
½) for trifluridine on Day 1 of Cycle 1 and on Day 12 of Cycle 1 were 1.4 hours and 2.1 hours, respectively. The mean t
½ values for tipiracil hydrochloride on Day 1 of Cycle 1 and on Day 12 of Cycle 1 were 2.1 hours and 2.4 hours, respectively.
Following a single dose of Lonsurf (35 mg/m
2) in patients with advanced solid tumours, the oral clearance (CL/F) for trifluridine and tipiracil hydrochloride were 10.5 L/hr and 109 L/hr, respectively. After single oral administration of Lonsurf with [
14C]-trifluridine, the total cumulative excretion of radioactivity was 60% of the administered dose. The majority of recovered radioactivity was eliminated into urine (55% of the dose) within 24 hours, and the excretion into faeces and expired air was less than 3% for both. After single oral administration of Lonsurf with [
14C]-tipiracil hydrochloride, recovered radioactivity was 77% of the dose, which consisted of 27% urinary excretion and 50% faecal excretion.
Linearity/non-linearity: In a dose finding study (15 to 35 mg/m
2 twice daily), the AUC from time 0 to 10 hours (AUC
0-10) of trifluridine tended to increase more than expected based on the increase in dose; however, oral clearance (CL/F) and apparent volume of distribution (Vd/F) of trifluridine were generally constant at the dose range of 20 to 35 mg/m
2. As for the other exposure parameters of trifluridine and tipiracil hydrochloride, those appeared to be dose proportional.
Pharmacokinetics in special populations: Age, gender and race: Based on the population PK analysis, there is no clinically relevant effect of age, gender or race on the PK of trifluridine or tipiracil hydrochloride.
Renal impairment: Of the 533 patients in the RECOURSE study who received Lonsurf, 306 (57%) patients had normal renal function (CrCl ≥ 90 mL/min), 178 (33%) patients had mild renal impairment (CrCl 60 to 89 mL/min), and 47 (9%) had moderate renal impairment (CrCl 30 to 59 mL/min), with data missing for 2 patients. Patients with severe renal impairment were not enrolled in the study.
Based on a population PK analysis, the exposure of Lonsurf in patients with mild renal impairment (CrCl = 60 to 89 mL/min) was similar to those in patients with normal renal function (CrCl ≥ 90 mL/min). A higher exposure of Lonsurf was observed in moderate renal impairment (CrCl = 30 to 59 mL/min). Estimated (CrCl) was a significant covariate for CL/F in both final models of trifluridine and tipiracil hydrochloride. The mean relative ratio of AUC in patients with mild (N = 38) and moderate (N = 16) renal impairment compared to patients with normal renal function (N = 84) were 1.31 and 1.43 for trifluridine, respectively, and 1.34 and 1.65 for tipiracil hydrochloride, respectively.
In a dedicated study the pharmacokinetics of trifluridine and tipiracil hydrochloride were evaluated in cancer patients with normal renal function (CrCl ≥ 90 mL/min, N = 12), mild renal impairment (CrCl = 60 to 89 mL/min, N = 12), moderate renal impairment (CrCl = 30 to 59 mL/min, N = 11), or severe renal impairment (CrCl = 15 to 29 mL/min, N = 8). Patients with severe renal impairment received an adjusted starting dose of 20 mg/m
2 twice daily (reduced to 15 mg/m
2 twice daily based on individual safety and tolerability). The effect of renal impairment after repeated administration was a 1.6- and 1.4-fold increase of trifluridine total exposure in patients with moderate and severe renal impairment, respectively, compared to patients with normal renal function; C
max remained similar. The total exposure of tipiracil hydrochloride in patients with moderate and severe renal impairment after repeated administration was 2.3- and 4.1-fold higher, respectively, compared to patients with normal renal function; this being linked to a more decreased clearance with increasing renal impairment. The PK of trifluridine and tipiracil hydrochloride have not been studied in patients with end-stage renal disease (CrCl < 15 mL/min or requiring dialysis) (see Dosage & Administration and Precautions).
Hepatic impairment: Based on the population PK analysis, liver function parameters including alkaline phosphatase (ALP, 36-2322 U/L), aspartate aminotransferase (AST, 11-197 U/L), alanine aminotransferase (ALT, 5-182 U/L), and total bilirubin (0.17-3.20 mg/dL) were not significant covariates for PK parameters of either trifluridine or tipiracil hydrochloride. Serum albumin was found to significantly affect trifluridine clearance, with a negative correlation. For low albumin values ranging from 2.2 to 3.5 g/dL, the corresponding clearance values range from 4.2 to 3.1 L/h.
In a dedicated study the PK of trifluridine and tipiracil hydrochloride were evaluated in cancer patients with mild or moderate hepatic impairment (National Cancer Institute [NCI] Criteria Group B and C, respectively) and in patients with normal hepatic function. Based upon limited data with a considerable variability, no statistically significant differences were observed in the pharmacokinetics in patients with normal hepatic function versus patients with mild or moderate hepatic impairment. No correlation was seen for trifluridine nor tipiracil hydrochloride between PK parameters and AST or/and total blood bilirubin. Half-life time (t
½) and the accumulation ratio of trifluridine and tipiracil hydrochloride were similar between the moderate, mild and normal hepatic function patients. There is no need for a starting dose adjustment in patients with mild hepatic impairment (see Dosage & Administration).
Gastrectomy: The influence of gastrectomy on PK parameters was not able to be examined in the population PK analysis because there were few patients who had undergone gastrectomy (1% of overall).
In vitro interaction studies: Trifluridine is a substrate of TPase, but is not metabolised by cytochrome P450 (CYP). Tipiracil hydrochloride is not metabolised in either human liver S9 or cryopreserved hepatocytes.
In vitro studies indicated that trifluridine, tipiracil hydrochloride and FTY (inactive metabolite of trifluridine) did not inhibit the CYP isoforms tested (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 and CYP3A4/5).
In vitro evaluation indicated that trifluridine, tipiracil hydrochloride and FTY had no inductive effect on human CYP1A2, CYP2B6 or CYP3A4/5. Thus, trifluridine and tipiracil hydrochloride are not expected to cause or be subject to a significant medicinal product interaction mediated by CYP.
In vitro evaluation of trifluridine and tipiracil hydrochloride was conducted using human uptake and efflux transporters (trifluridine with MDR1, OATP1B1, OATP1B3 and BCRP; tipiracil hydrochloride with OAT1, OAT3, OCT2, MATE1, MDR1 and BCRP). Neither trifluridine nor tipiracil hydrochloride was an inhibitor of or substrate for human uptake and efflux transporters based on
in vitro studies, except for OCT2 and MATE1. Tipiracil hydrochloride was an inhibitor of OCT2 and MATE1
in vitro, but at concentrations substantially higher than human plasma C
max at steady state. Thus, it is unlikely to cause an interaction with other medicinal products, at recommended doses, due to inhibition of OCT2 and MATE1. Transport of tipiracil hydrochloride by OCT2 and MATE1 might be affected when Lonsurf is administered concomitantly with inhibitors of OCT2 and MATE1.
Pharmacokinetic/pharmacodynamic relationship: The efficacy and safety of Lonsurf in metastatic colorectal cancer was compared between a high-exposure group (> median) and a low-exposure group (≤ median) based on the median AUC value of trifluridine. OS appeared more favourable in the high AUC group compared to the low AUC group (median OS of 9.3 vs. 8.1 months, respectively). All AUC groups performed better than placebo throughout the follow-up period. The incidences of Grade ≥ 3 neutropenia were higher in the high-trifluridine AUC group (47.8%) compared with the low-trifluridine AUC group (30.4%).
Toxicology: Preclinical safety data: Repeat-dose toxicity: Toxicology assessment of trifluridine/tipiracil hydrochloride was performed in rats, dogs and monkeys. The target organs identified were the lymphatic and haematopoietic systems and the gastrointestinal tract. All changes, i.e., leukopenia, anaemia, bone marrow hypoplasia, atrophic changes in the lymphatic and haematopoietic tissues and the gastrointestinal tract, were reversible within 9 weeks of drug withdrawal. Whitening, breakage, and malocclusion were observed in teeth of rats treated with trifluridine/tipiracil hydrochloride, which are considered rodent specific and not relevant for human.
Carcinogenesis and mutagenesis: No long term studies evaluating the carcinogenic potential of trifluridine/tipiracil hydrochloride in animals have been performed. Trifluridine was shown to be genotoxic in a reverse mutation test in bacteria, a chromosomal aberration test in mammal-cultured cells, and a micronucleus test in mice. Therefore, Lonsurf should be treated as a potential carcinogen.
Reproductive toxicity: Results of animal studies did not indicate an effect of trifluridine and tipiracil hydrochloride on male and female fertility in rats. The increases in the corpus luteum count and implanting embryo count observed in female rats at high doses were not considered adverse (see Use in Pregnancy & Lactation). Lonsurf has been shown to cause embryo-foetal lethality and embryo-foetal toxicity in pregnant rats when given at dose levels lower than the clinical exposure. No peri/post-natal developmental toxicity studies have been performed.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image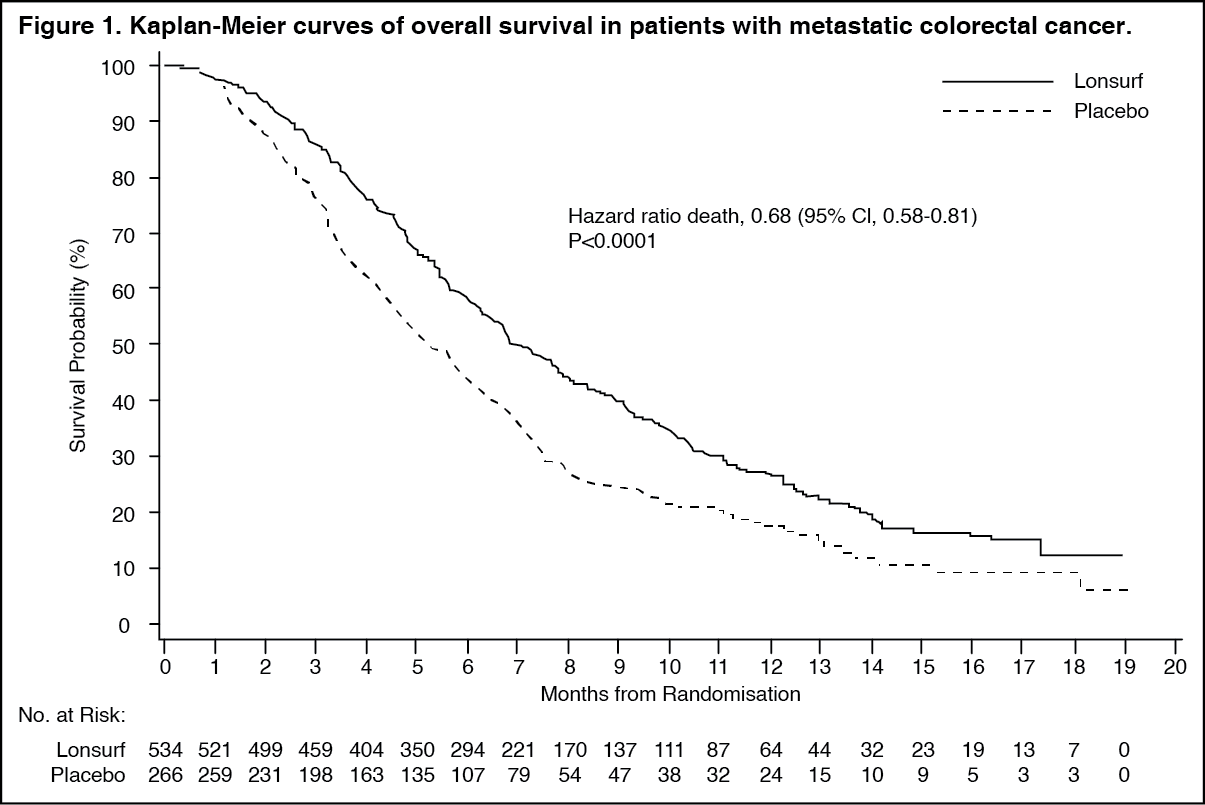 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image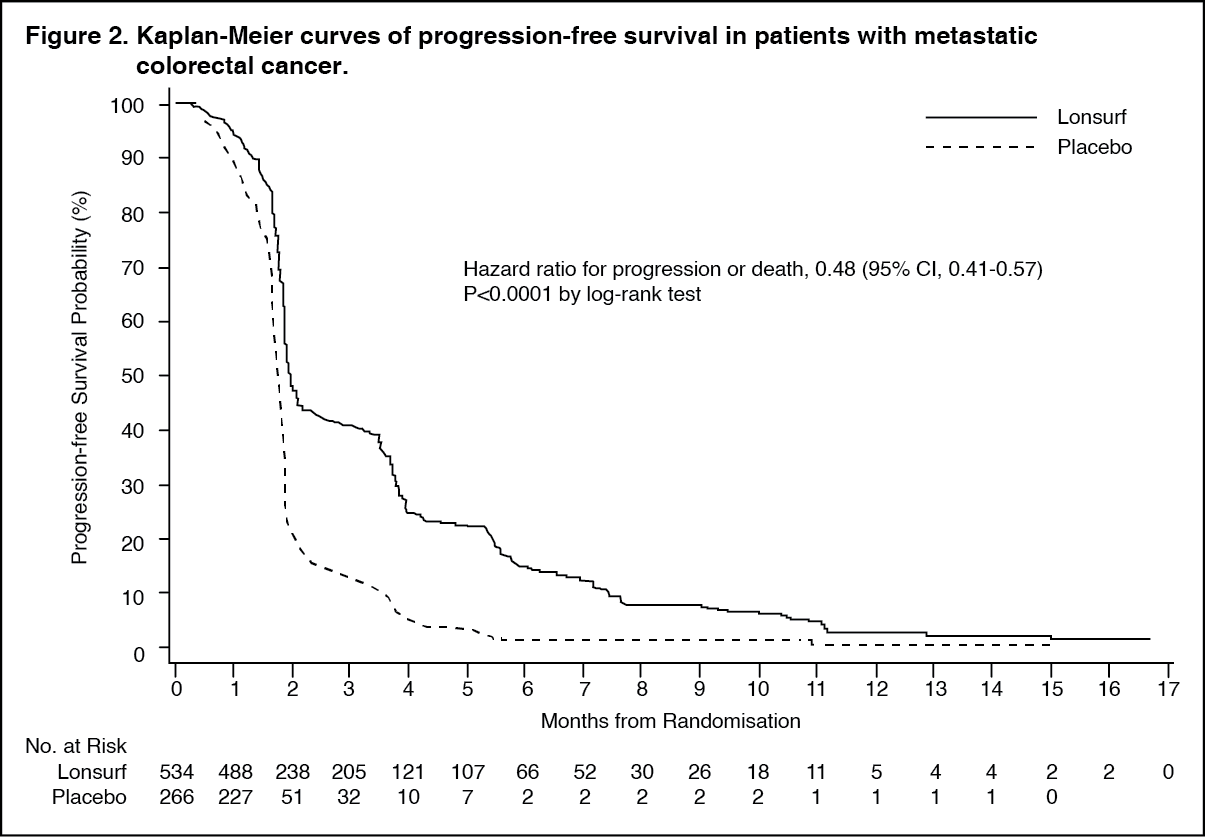 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image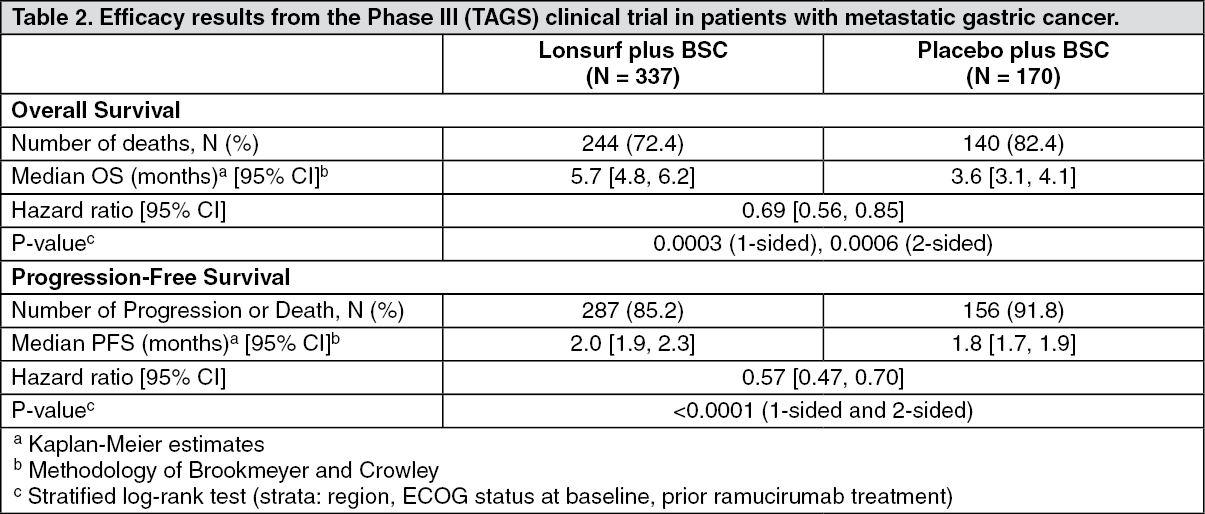 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image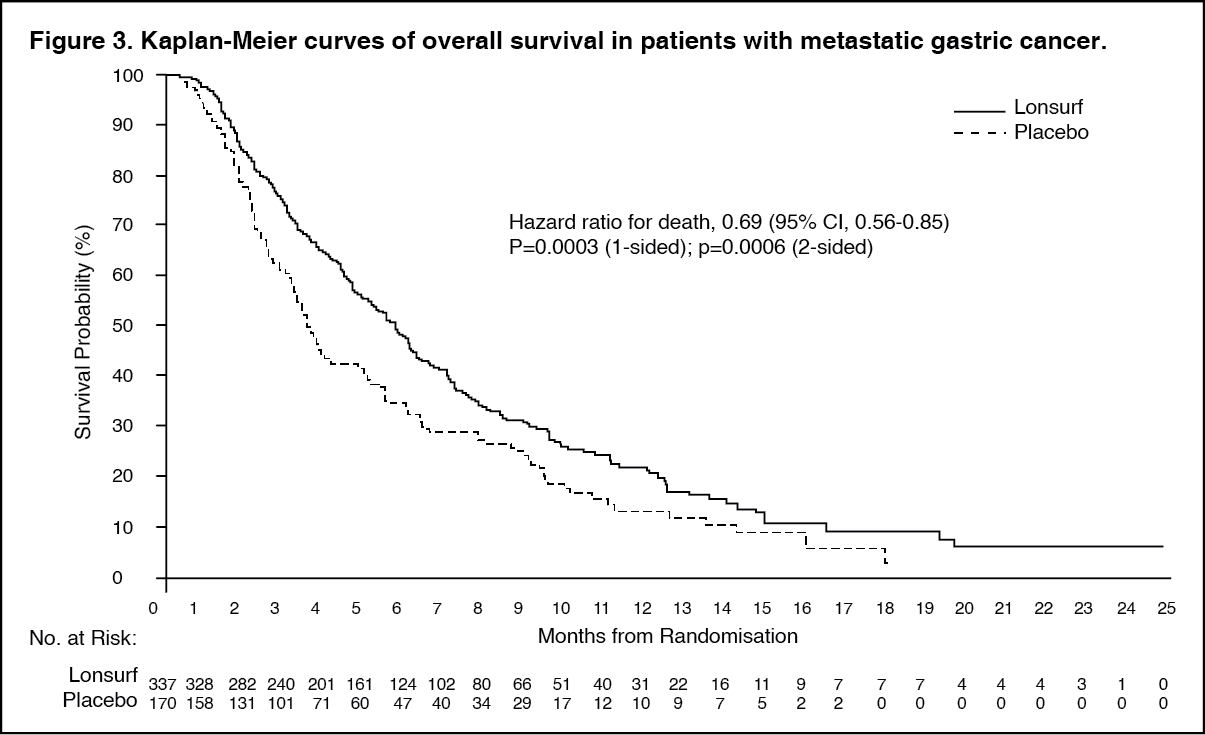 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image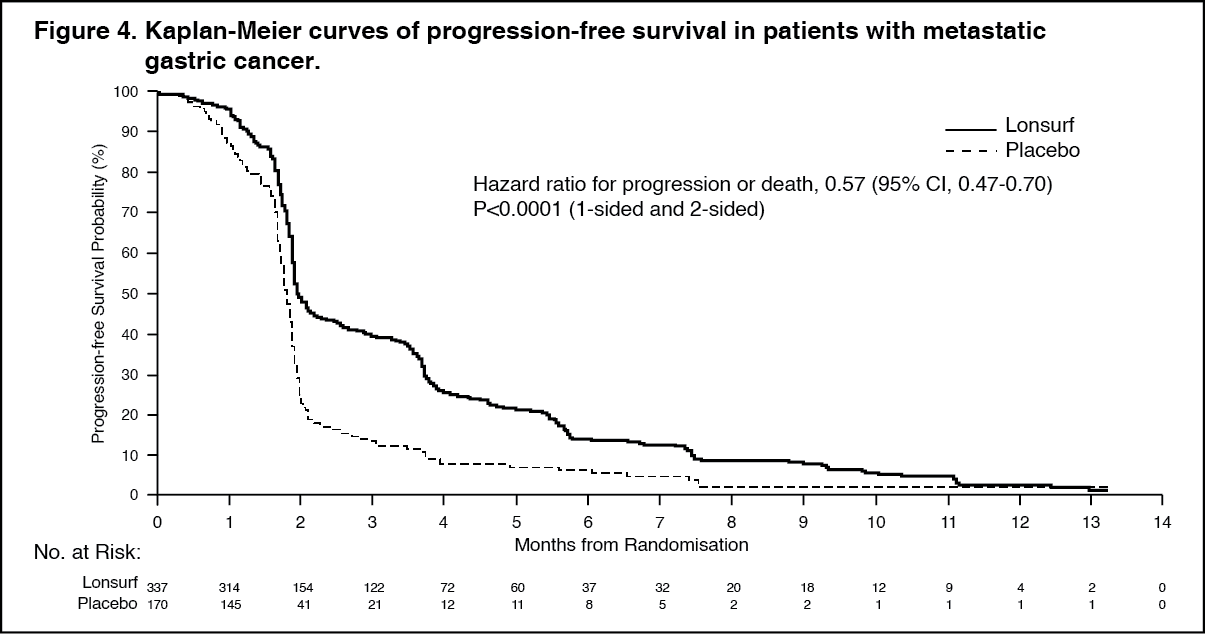 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image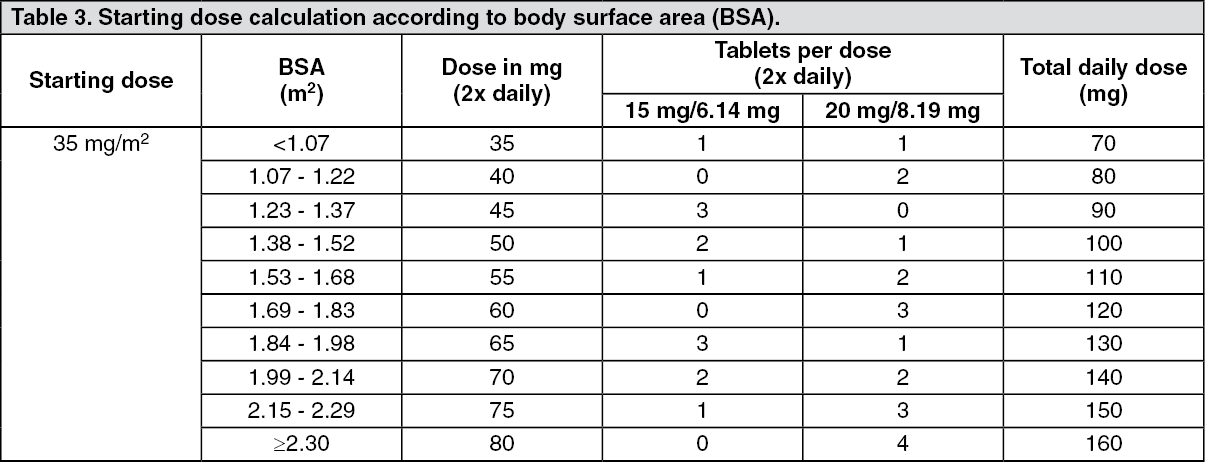 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image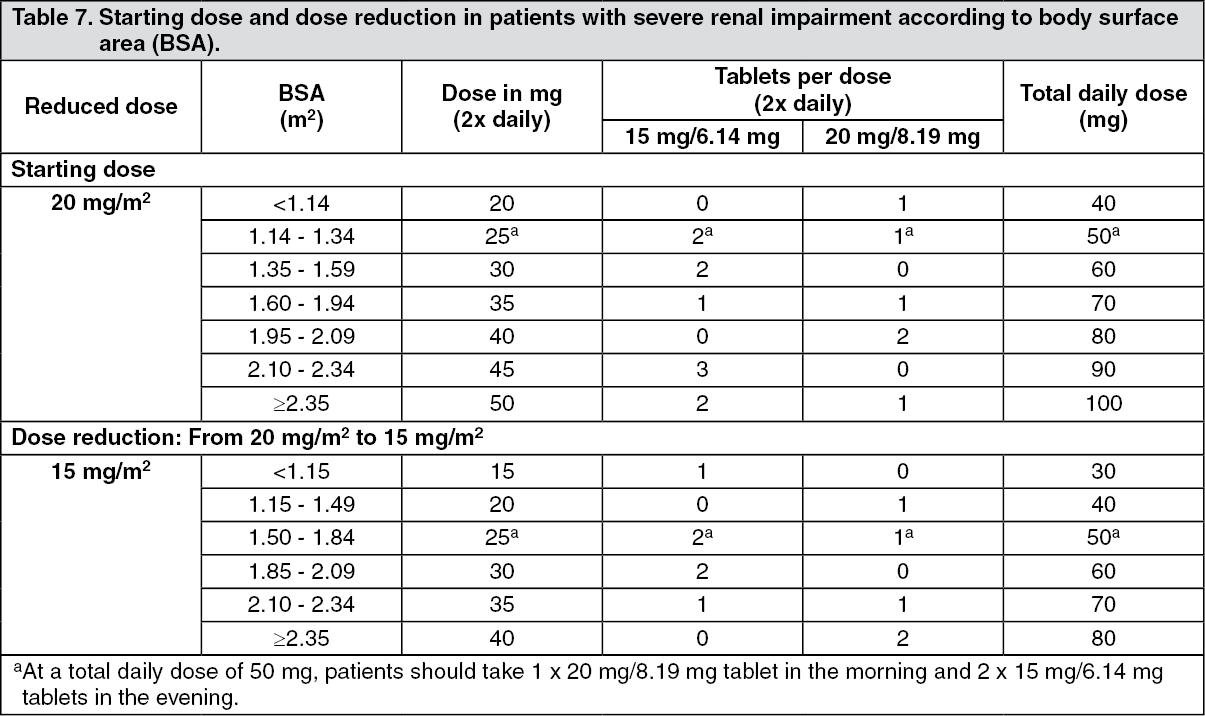 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image


 Sign Out
Sign Out