


 Sign Out
Sign Out

Alogliptin is a DPP-4 inhibitor that slows the inactivation of the incretin hormones, thereby increasing their bloodstream concentrations and reducing fasting and postprandial glucose concentrations in a glucose-dependent manner in patients with type 2 diabetes mellitus. Alogliptin selectively binds to and inhibits DPP-4 but not DPP-8 or DPP-9 activity in vitro at concentrations approximating therapeutic exposures.
Pharmacodynamics: Single-dose administration of NESINA to healthy subjects resulted in a peak inhibition of DPP-4 within 2 to 3 hours after dosing. The peak inhibition of DPP-4 exceeded 93% across doses of 12.5 mg to 800 mg. Inhibition of DPP-4 remained above 80% at 24 hours for doses greater than or equal to 25 mg. Peak and total exposure over 24 hours to active GLP-1 were 3- to 4-fold greater with NESINA (at doses of 25 to 200 mg) than placebo. In a 16-week, double-blind, placebo-controlled study, NESINA 25 mg demonstrated decreases in postprandial glucagon while increasing postprandial active GLP-1 levels compared to placebo over an 8-hour period following a standardized meal. It is unclear how these findings relate to changes in overall glycemic control in patients with type 2 diabetes mellitus. In this study, NESINA 25 mg demonstrated decreases in 2-hour postprandial glucose compared to placebo (-30 mg/dL versus 17 mg/dL, respectively).
Multiple-dose administration of alogliptin to patients with type 2 diabetes also resulted in a peak inhibition of DPP-4 within 1 to 2 hours and exceeded 93% across all doses (25 mg, 100 mg and 400 mg) after a single dose and after 14 days of once-daily dosing. At these doses of NESINA, inhibition of DPP-4 remained above 81% at 24 hours after 14 days of dosing.
Cardiac Electrophysiology: In a randomized, placebo-controlled, 4-arm, parallel-group study, 257 subjects were administered either alogliptin 50 mg, alogliptin 400 mg, moxifloxacin 400 mg or placebo once- daily for a total of 7 days. No increase in corrected QT (QTc) was observed with either dose of alogliptin. At the 400 mg dose, peak alogliptin plasma concentrations were 19-fold higher than the peak concentrations following the maximum recommended clinical dose of 25 mg.
Clinical Studies: NESINA has been studied as monotherapy and in combination with metformin, a sulfonylurea, a thiazolidinedione (either alone or in combination with metformin or a sulfonylurea), and insulin (either alone or in combination with metformin).
A total of 14,053 patients with type 2 diabetes were randomized in 11 double-blind, placebo- or active-controlled clinical safety and efficacy studies conducted to evaluate the effects of NESINA on glycemic control. The racial distribution of patients exposed to study medication was 70% Caucasian, 17% Asian, 6% Black, and 7% other racial groups. The ethnic distribution was 30% Hispanic. Patients had an overall mean age of 57 years (range 21 to 91 years).
In patients with type 2 diabetes, treatment with NESINA produced clinically meaningful and statistically significant improvements in A1C compared to placebo. As is typical for trials of agents to treat type 2 diabetes, the mean reduction in A1C with NESINA appears to be related to the degree of A1C elevation at baseline.
NESINA had similar changes from baseline in serum lipids compared to placebo.
Patients with Inadequate Glycemic Control on Diet and Exercise: A total of 1768 patients with type 2 diabetes participated in three double-blind studies to evaluate the efficacy and safety of NESINA in patients with inadequate glycemic control on diet and exercise. All three studies had a 4-week, single-blind, placebo run-in period followed by a 26-week randomized treatment period.
Patients who failed to meet prespecified hyperglycemic goals during the 26-week treatment periods received glycemic rescue therapy.
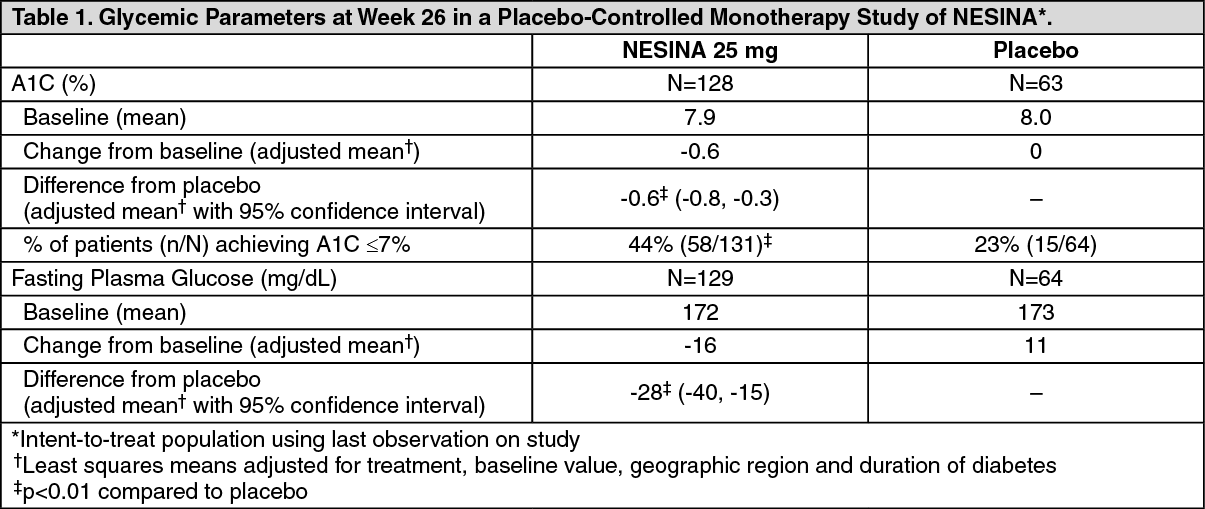
In a 26-week, double-blind, placebo-controlled study, a total of 329 patients (mean baseline A1C = 8%) were randomized to receive NESINA 12.5 mg, NESINA 25 mg or placebo once daily. Treatment with NESINA 25 mg resulted in statistically significant improvements from baseline in A1C and fasting plasma glucose (FPG) compared to placebo at Week 26 (Table 1). A total of 8% of patients receiving NESINA 25 mg and 30% of those receiving placebo required glycemic rescue therapy.
Improvements in A1C were not affected by gender, age, or baseline body mass index (BMI).
The mean change in body weight with NESINA was similar to placebo. (See Table 1.)
 Click on icon to see table/diagram/image
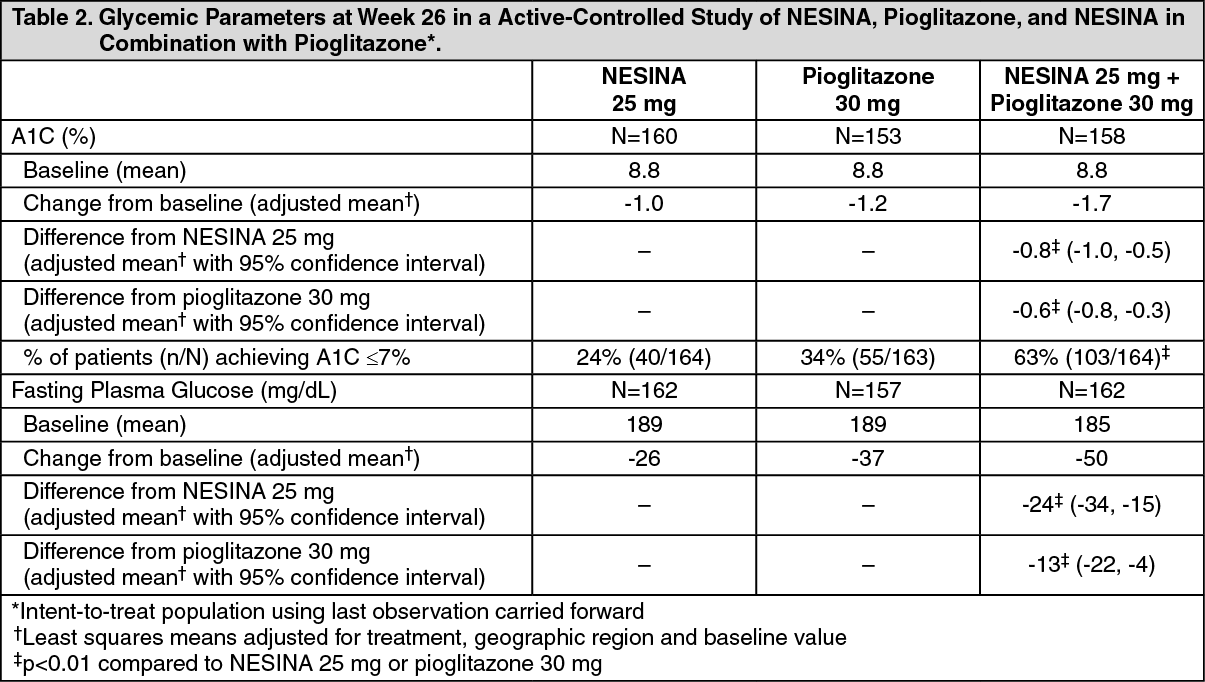
Click on icon to see table/diagram/imageIn a 26-week, double-blind, active-controlled study, a total of 655 patients (mean baseline A1C = 8.8%) were randomized to receive NESINA 25 mg alone, pioglitazone 30 mg alone, NESINA 12.5 mg with pioglitazone 30 mg or NESINA 25 mg with pioglitazone 30 mg once daily. Coadministration of NESINA 25 mg with pioglitazone 30 mg resulted in statistically significant improvements from baseline in A1C and FPG compared to NESINA 25 mg alone and to pioglitazone 30 mg alone (Table 2). A total of 3% of patients receiving NESINA 25 mg coadministered with pioglitazone 30 mg, 11% of those receiving NESINA 25 mg alone and 6% of those receiving pioglitazone 30 mg alone required glycemic rescue.
Improvements in A1C were not affected by gender, age or baseline BMI.
The mean increase in body weight was similar between pioglitazone alone and NESINA when coadministered with pioglitazone. (See Table 2.)
 Click on icon to see table/diagram/image
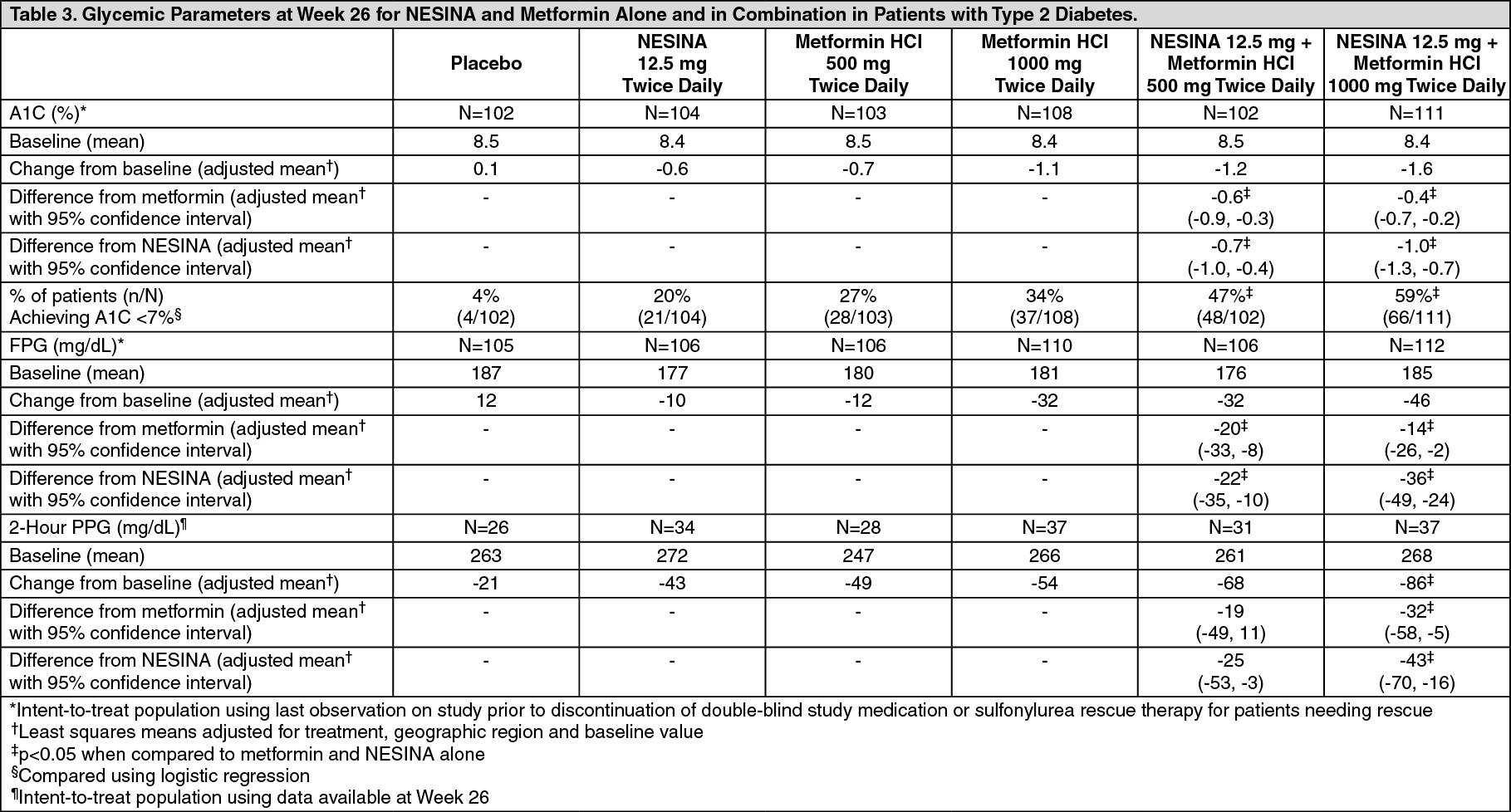
Click on icon to see table/diagram/imageIn a 26-week, double-blind, placebo-controlled study, a total of 784 patients inadequately controlled on diet and exercise alone (mean baseline A1C = 8.4%) were randomized to 1 of 7 treatment groups: placebo; metformin HCl 500 mg or metformin HCl 1000 mg twice daily; NESINA 12.5 mg twice daily; NESINA 25 mg daily; or NESINA 12.5 mg in combination with metformin HCl 500 mg or metformin HCl 1000 mg twice daily.
Both coadministration treatment arms (NESINA 12.5 mg + metformin HCl 500 mg and NESINA 12.5 mg + metformin HCl 1000 mg) resulted in statistically significant improvements in A1C and FPG when compared with their respective individual alogliptin and metformin component regimens (Table 3). Coadministration treatment arms demonstrated improvements in two-hour postprandial glucose (PPG) compared to NESINA alone or metformin alone (Table 3). A total of 12.3% of patients receiving NESINA 12.5 mg + metformin HCl 500 mg, 2.6% of patients receiving NESINA 12.5 mg + metformin HCl 1000 mg, 17.3% of patients receiving NESINA 12.5 mg, 22.9% of patients receiving metformin HCl 500 mg, 10.8% of patients receiving metformin HCl 1000 mg and 38.7% of patients receiving placebo required glycemic rescue.
Improvements in A1C were not affected by gender, age, race, or baseline BMI. The mean decrease in body weight was similar between metformin alone and NESINA when coadministered with metformin. (See Table 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageCombination Therapy: Add-On Therapy as inadequately controlled on metformin at the maximum tolerated dose: A total of 2081 patients with type 2 diabetes participated in two 26-week, double-blind, placebo-controlled studies to evaluate the efficacy and safety of NESINA as add-on therapy to metformin. In both studies, patients were inadequately controlled on metformin at a dose of at least 1500 mg per day or at the maximum tolerated dose. All patients entered a 4-week, single-blind placebo run-in period prior to randomization. Patients who failed to meet prespecified hyperglycemic goals during the 26-week treatment periods received glycemic rescue therapy.
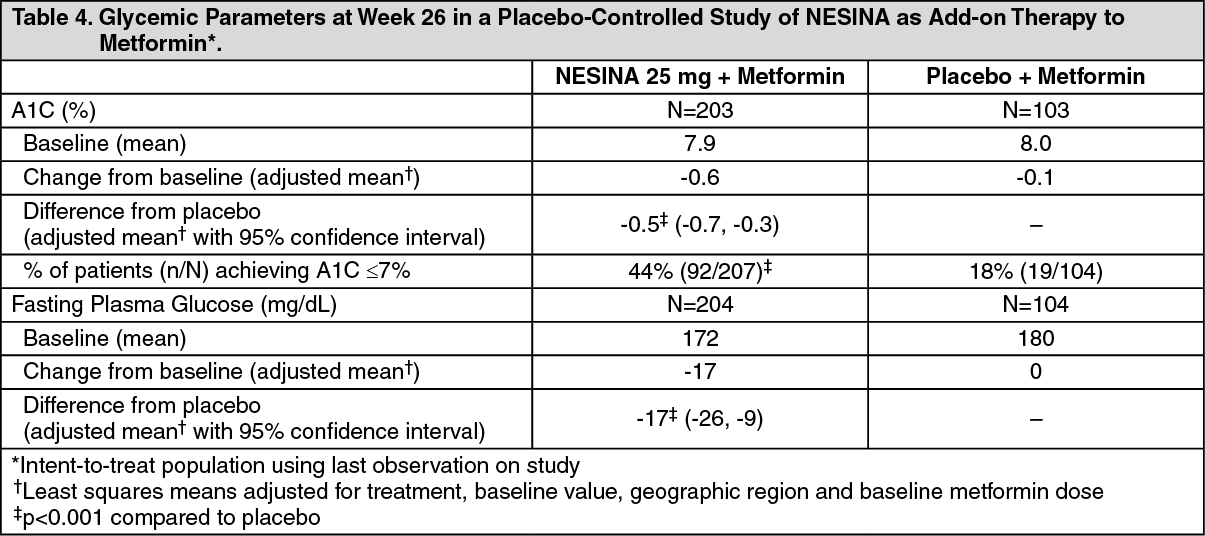
In the first 26-week, placebo-controlled study, a total of 527 patients already on metformin (mean baseline A1C = 8%) were randomized to receive NESINA 12.5 mg, NESINA 25 mg or placebo. Patients were maintained on a stable dose of metformin (median dose = 1700 mg) during the treatment period. NESINA 25 mg in combination with metformin resulted in statistically significant improvements from baseline in A1C and FPG at Week 26, when compared to placebo (Table 4). A total of 8% of patients receiving NESINA 25 mg and 24% of patients receiving placebo required glycemic rescue.
Improvements in A1C were not affected by gender, age, baseline BMI, or baseline metformin dose. The mean decrease in body weight was similar between NESINA and placebo when given in combination with metformin. (See Table 4.)
 Click on icon to see table/diagram/image
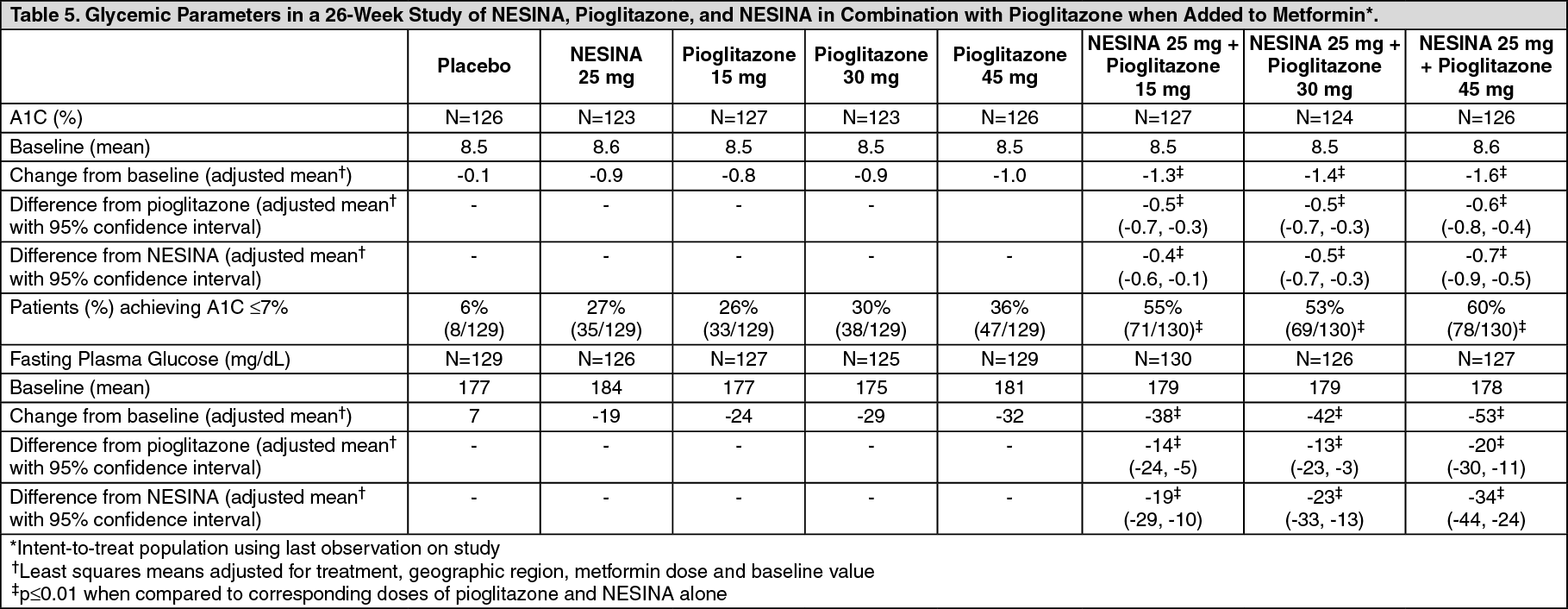
Click on icon to see table/diagram/imageIn the second 26-week double-blind, placebo-controlled study, a total of 1554 patients already on metformin (mean baseline A1C = 8.5%) were randomized to one of 12 double-blind treatment groups: placebo; 12.5 mg or 25 mg of NESINA alone; 15 mg, 30 mg or 45 mg of pioglitazone alone; or 12.5 mg or 25 mg of NESINA in combination with 15 mg, 30 mg, or 45 mg of pioglitazone. Patients were maintained on a stable dose of metformin (median dose = 1700 mg) during the treatment period. Coadministration of NESINA and pioglitazone provided statistically significant improvements in A1C and FPG compared to placebo, to NESINA alone or to pioglitazone alone when added to background metformin therapy (Table 5, Figure 1). In addition, improvements from baseline A1C were comparable between NESINA alone and pioglitazone alone (15 mg, 30 mg, and 45 mg) at Week 26. A total of 4%, 5%, or 2% of patients receiving NESINA 25 mg with 15 mg, 30 mg, or 45 mg pioglitazone, 33% of patients receiving placebo, 13% of patients receiving NESINA 25 mg and 10%, 15%, or 9% of patients receiving pioglitazone 15 mg, 30 mg, or 45 mg alone required glycemic rescue.
Improvements in A1C were not affected by gender, age or baseline BMI.
The mean increase in body weight was similar between pioglitazone alone and NESINA when coadministered with pioglitazone. (See Table 5 and Figure 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image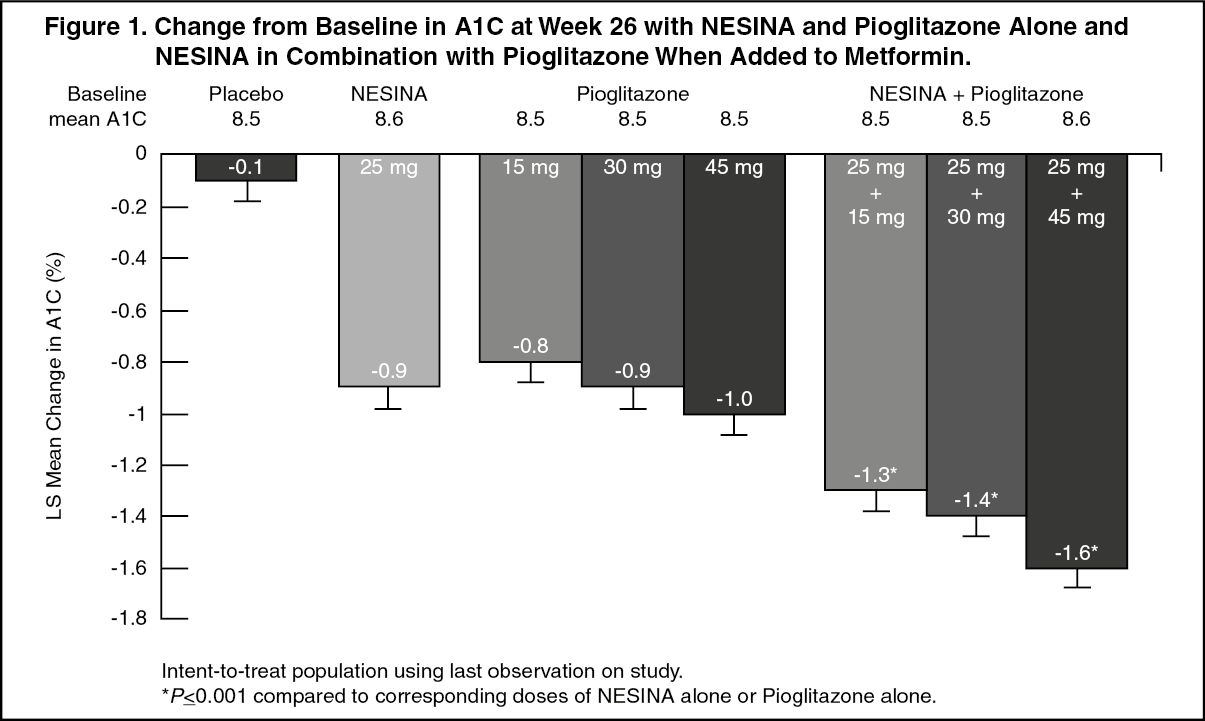 Click on icon to see table/diagram/image
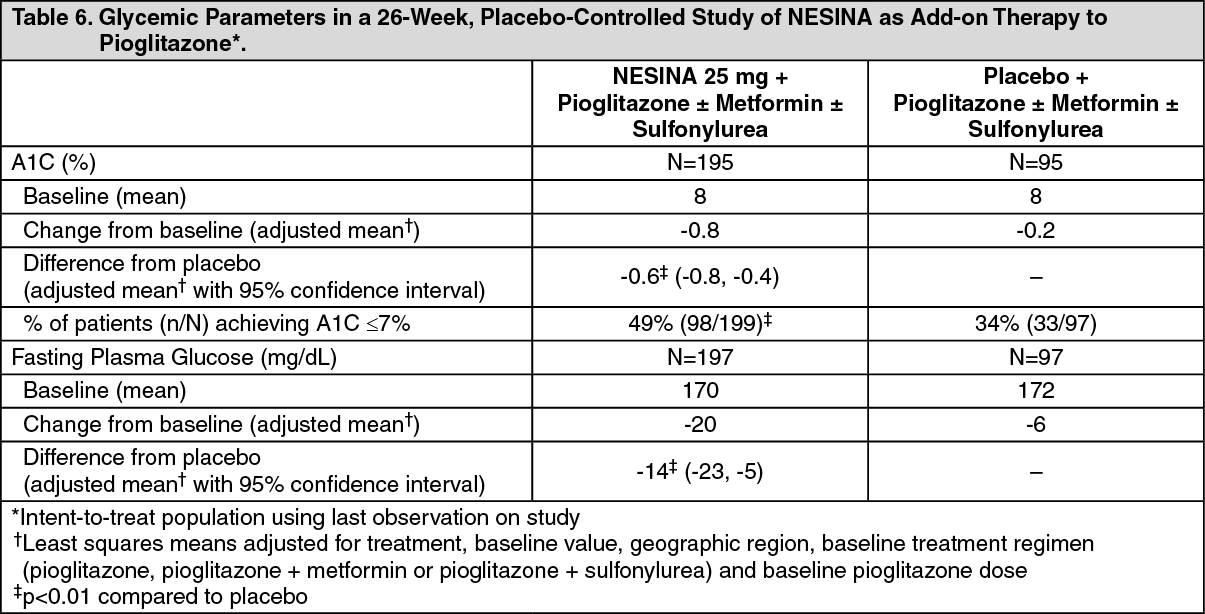
Click on icon to see table/diagram/imageAdd-On Therapy to a Thiazolidinedione: In a 26-week, placebo-controlled study, a total of 493 patients inadequately controlled on a thiazolidinedione alone or in combination with metformin or a sulfonylurea (10 mg) (mean baseline A1C = 8%) were randomized to receive NESINA 12.5 mg, NESINA 25 mg or placebo. Patients were maintained on a stable dose of pioglitazone (median dose = 30 mg) during the treatment period; those who were also previously treated on metformin (median dose = 2000 mg) or sulfonylurea (median dose = 10 mg) prior to randomization were maintained on the combination therapy during the treatment period. All patients entered into a 4-week, single-blind placebo run-in period prior to randomization. Patients who failed to meet pre-specified hyperglycemic goals during the 26-week treatment period received glycemic rescue therapy.
The addition of NESINA 25 mg once daily to pioglitazone therapy resulted in statistically significant improvements from baseline in A1C and FPG at Week 26, compared to placebo (Table 6). A total of 9% of patients who were receiving NESINA 25 mg and 12% of patients receiving placebo required glycemic rescue.
Improvements in A1C were not affected by gender, age, baseline BMI, or baseline pioglitazone dose.
Clinically meaningful reductions in A1C were observed with NESINA compared to placebo regardless of whether subjects were receiving concomitant metformin or sulfonylurea (-0.2% placebo versus -0.9% NESINA) therapy or pioglitazone alone (0% placebo versus -0.52% NESINA).
The mean increase in body weight was similar between NESINA and placebo when given in combination with pioglitazone. (See Table 6.)
 Click on icon to see table/diagram/image
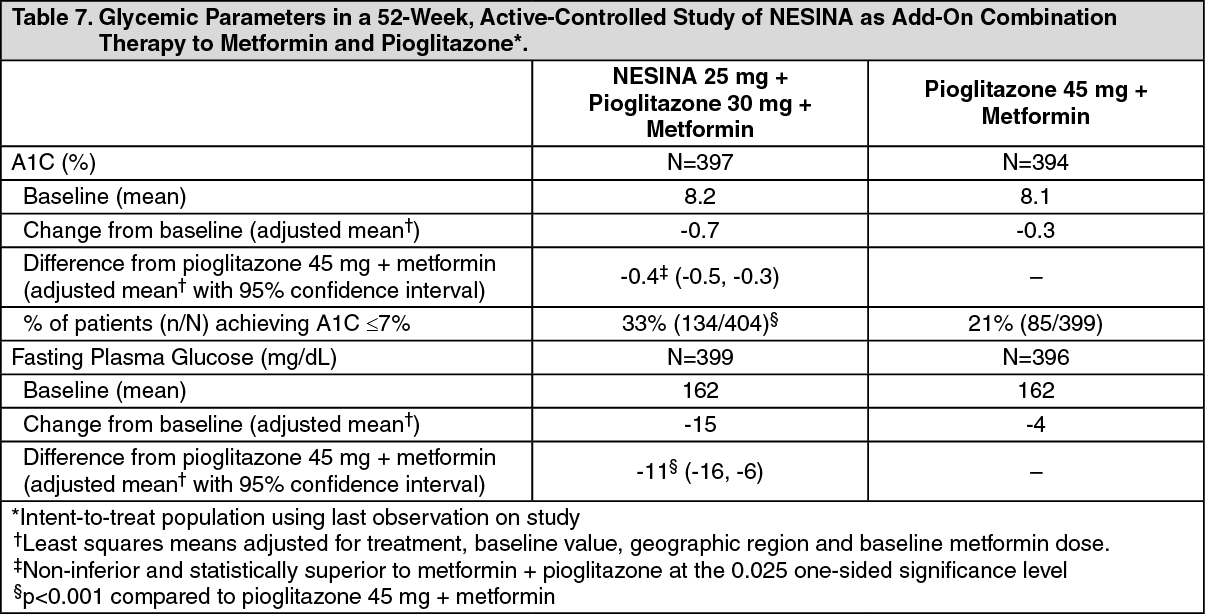
Click on icon to see table/diagram/imageAdd-on Combination Therapy with Pioglitazone and Metformin: In a 52-week, active-comparator study, a total of 803 patients inadequately controlled (mean baseline A1C = 8.2%) on a current regimen of pioglitazone 30 mg and metformin at least 1500 mg per day or at the maximum tolerated dose were randomized to either receive the addition of NESINA 25 mg or the titration of pioglitazone 30 mg to 45 mg following a 4-week, single-blind placebo run-in period. Patients were maintained on a stable dose of metformin (median dose = 1700 mg). Patients who failed to meet prespecified hyperglycemic goals during the 52-week treatment period received glycemic rescue therapy.
In combination with pioglitazone and metformin, NESINA 25 mg was shown to be statistically superior in lowering A1C and FPG compared with the titration of pioglitazone from 30 mg to 45 mg at Week 26 and at Week 52 (Table 7; results shown only for Week 52). A total of 11% of patients in the NESINA 25 mg treatment group and 22% of patients in the pioglitazone up-titration group required glycemic rescue.
Improvements in A1C were not affected by gender, age, race or baseline BMI.
The mean increase in body weight was similar in both treatment arms. (See Table 7.)
 Click on icon to see table/diagram/image
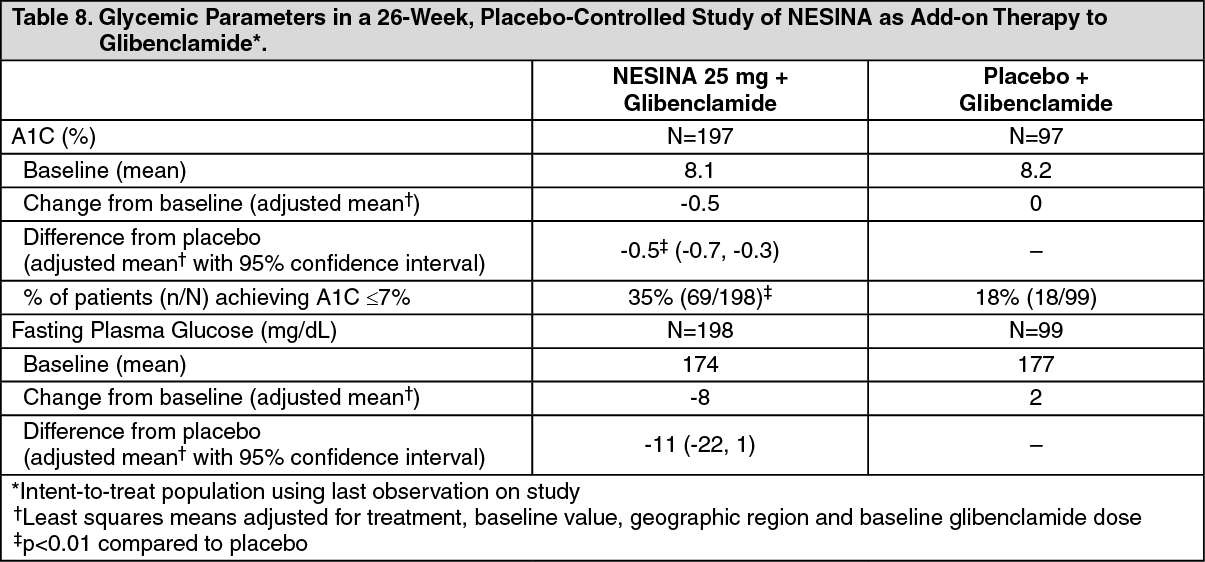
Click on icon to see table/diagram/imageAdd-On Therapy to a Sulfonylurea: In a 26-week, placebo-controlled study, a total of 500 patients inadequately controlled on a sulfonylurea (mean baseline A1C = 8.1%) were randomized to receive NESINA 12.5 mg, NESINA 25 mg or placebo. Patients were maintained on a stable dose of glibenclamide (median dose = 10 mg) during the treatment period. All patients entered into a four-week, single-blind, placebo run-in period prior to randomization. Patients who failed to meet pre-specified hyperglycemic goals during the 26-week treatment period received glycemic rescue therapy.
The addition of NESINA 25 mg to glibenclamide therapy resulted in statistically significant improvements from baseline in A1C at Week 26 when compared to placebo (Table 8). Improvements in FPG observed with NESINA 25 mg were not statistically significant compared with placebo. A total of 16% of patients receiving NESINA 25 mg and 28% of those receiving placebo required glycemic rescue.
Improvements in A1C were not affected by gender, age, baseline BMI or baseline glibenclamide dose. The mean change in body weight was similar between NESINA and placebo when given in combination with glibenclamide. (See Table 8.)
 Click on icon to see table/diagram/image
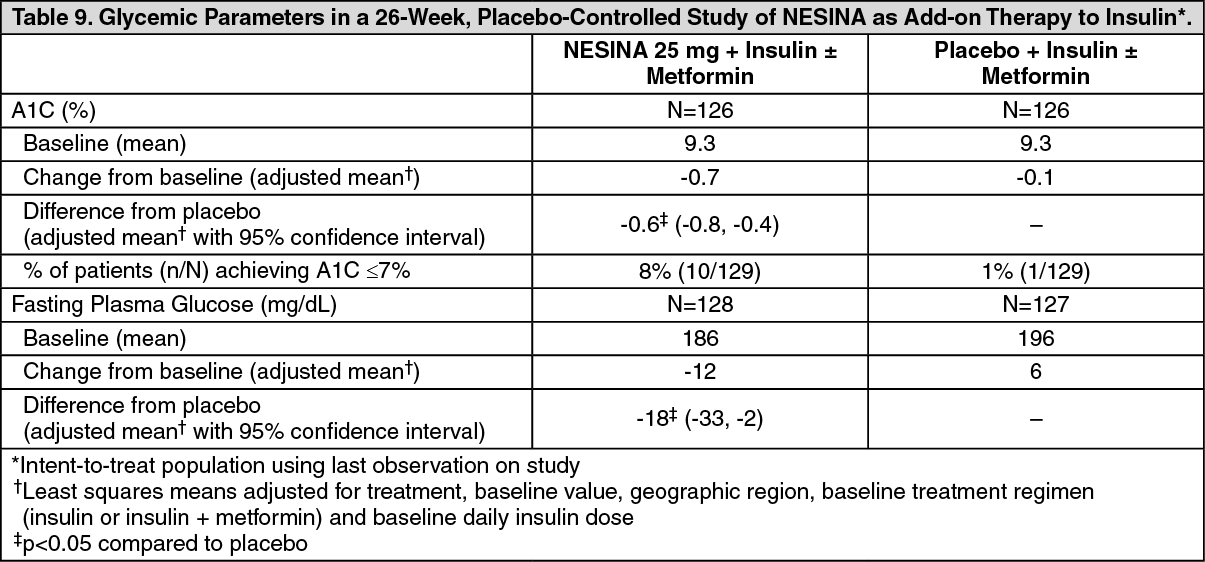
Click on icon to see table/diagram/imageAdd-On Therapy to Insulin: In a 26-week, placebo-controlled study, a total of 390 patients inadequately controlled on insulin alone (42%) or in combination with metformin (58%) (mean baseline A1C = 9.3%) were randomized to receive NESINA 12.5 mg, NESINA 25 mg or placebo. Patients were maintained on their insulin regimen (median dose = 55 IU) upon randomization and those previously treated with insulin in combination with metformin (median dose = 1700 mg) prior to randomization continued on the combination regimen during the treatment period. Patients entered the trial on short-, intermediate- or long-acting (basal) insulin or premixed insulin. Patients who failed to meet pre-specified hyperglycemic goals during the 26-week treatment period received glycemic rescue therapy.
The addition of NESINA 25 mg once daily to insulin therapy resulted in statistically significant improvements from baseline in A1C and FPG at Week 26, when compared to placebo (Table 9). A total of 20% of patients receiving NESINA 25 mg and 40% of those receiving placebo required glycemic rescue.
Improvements in A1C were not affected by gender, age, baseline BMI or baseline insulin dose. Clinically meaningful reductions in A1C were observed with NESINA compared to placebo regardless of whether subjects were receiving concomitant metformin and insulin (-0.2% placebo versus -0.8% NESINA) therapy or insulin alone (0.1% placebo versus -0.7% NESINA). (See Table 9.)
The mean increase in body weight was similar between NESINA and placebo when given in combination with insulin.
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageCardiovascular Safety Trial: A randomized, double-blind, placebo-controlled cardiovascular outcomes trial (EXAMINE) was conducted to evaluate the cardiovascular risk of NESINA. The trial compared the risk of major adverse cardiovascular events (MACE) between NESINA (N=2701) and placebo (N=2679) when added to standard of care therapies for diabetes and atherosclerotic vascular disease (ASCVD). The trial was event driven and patients were followed until a sufficient number of primary outcome events accrued.
Eligible patients were adults with type 2 diabetes who had inadequate glycemic control at baseline (e.g., HbA1c >6.5%) and had been hospitalized for an acute coronary syndrome event (e.g., acute myocardial infarction or unstable angina requiring hospitalization) 15 to 90 days prior to randomization. The dose of NESINA was based on estimated renal function at baseline per dosage and administration recommendations (see Dosage and Administration). The average time between an acute coronary syndrome event and randomization was approximately 48 days.
The mean age of the population was 61 years. Most patients were male (68%), Caucasian (73%), and were recruited from outside of the United States (86%). Asian and Black patients contributed 20% and 4% of the total population, respectively. At the time of randomization patients had a diagnosis of type 2 diabetes mellitus for approximately 9 years, 87% had a prior myocardial infarction and 14% were current smokers. Hypertension (83%) and renal impairment (27% with an eGFR ≤60 ml/min/1.73 m2) were prevalent co-morbid conditions. Use of medications to treat diabetes (e.g., metformin 73%, sulfonylurea 54%, insulin 41%), and ASCVD (e.g., statin 94%, aspirin 93%, renin angiotensin system blocker 88%, beta-blocker 87%) was similar between patients randomized to NESINA and placebo at baseline. During the trial, medications to treat diabetes and ASCVD could be adjusted to ensure care for these conditions adhered to standard of care recommendations set by local practice guidelines.
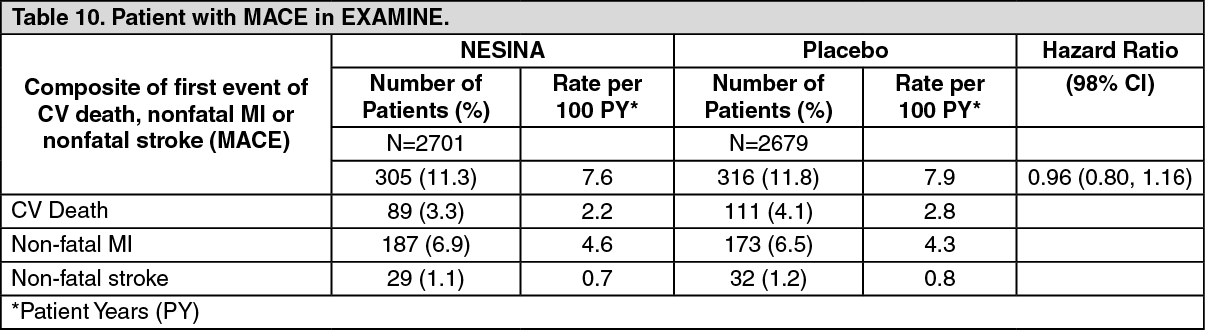
The primary endpoint in EXAMINE was the time to first occurrence of a MACE defined as the composite of cardiovascular death, nonfatal myocardial infarction (MI), or nonfatal stroke. The study was designed to exclude a pre-specified risk margin of 1.3 for the hazard ratio of MACE. The median exposure to study drug was 526 days and 95% of the patients were followed to study completion or death.
Table 10 shows the study results for the primary MACE composite endpoint and the contribution of each component to the primary MACE endpoint. The upper bound of the confidence interval was 1.16 and excluded a risk margin larger than 1.3. (See Table 10.)
 Click on icon to see table/diagram/image
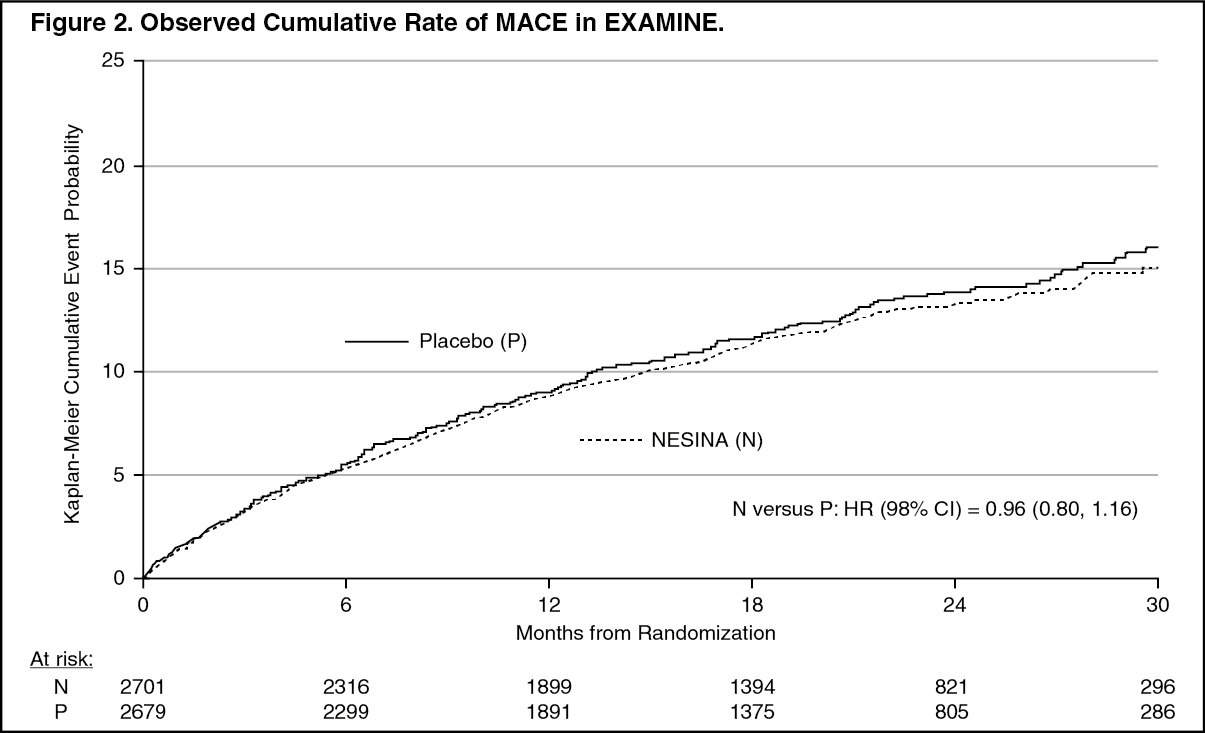
Click on icon to see table/diagram/imageThe Kaplan-Meier based cumulative event probability is presented in Figure 2 for the time to first occurrence of the primary MACE composite endpoint by treatment arm. The curves for placebo and NESINA overlap throughout the duration of the study. The observed incidence of MACE was highest within the first 60 days after randomization in both treatment arms (14.8 MACE per 100 PY), decreased from day 60 to the end of the first year (8.4 per 100 PY) and was lowest after 1 year of follow-up (5.2 per 100 PY). (See Figure 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe rate of all cause death was similar between treatment arms with 153 (3.6 per 100 PY) recorded among patients randomized to NESINA and 173 (4.1 per 100 PY) among patients randomized to placebo. A total of 112 deaths (2.9 per 100 PY) among patients on NESINA and 130 among patients on placebo (3.5 per 100PY) were adjudicated as cardiovascular deaths.
Pharmacokinetics: The pharmacokinetics of NESINA has been studied in healthy subjects and in patients with type 2 diabetes. After administration of single, oral doses up to 800 mg in healthy subjects, the peak plasma alogliptin concentration (median Tmax) occurred one to two hours after dosing. At the maximum recommended clinical dose of 25 mg, NESINA was eliminated with a mean terminal half-life (T1/2) of approximately 21 hours.
After multiple-dose administration up to 400 mg for 14 days in patients with type 2 diabetes, accumulation of alogliptin was minimal with an increase in total [e.g., area under the plasma concentration curve (AUC)] and peak (i.e., Cmax) alogliptin exposures of 34% and 9%, respectively. Total and peak exposure to alogliptin increased proportionally across single doses and multiple doses of alogliptin ranging from 25 mg to 400 mg. The intersubject coefficient of variation for alogliptin AUC was 17%. The pharmacokinetics of NESINA was also shown to be similar in healthy subjects and in patients with type 2 diabetes.
Absorption: The absolute oral bioavailability of NESINA is approximately 100%. Administration of NESINA with a high-fat meal results in no significant change in total and peak exposure to alogliptin. NESINA may therefore be administered with or without food.
Distribution: Following a single, 12.5 mg intravenous infusion of alogliptin to healthy subjects, the volume of distribution during the terminal phase was 417 L, indicating that the drug is well distributed into tissues.
Alogliptin is 20% bound to plasma proteins.
Metabolism: Alogliptin does not undergo extensive metabolism and 60% to 71% of the dose is excreted as unchanged drug in the urine.
Two minor metabolites were detected following administration of an oral dose of [14C] alogliptin, N-demethylated, M-I (less than 1% of the parent compound), and N-acetylated alogliptin, M-II (less than 6% of the parent compound). M-I is an active metabolite and is an inhibitor of DPP-4 similar to the parent molecule; M-II does not display any inhibitory activity toward DPP-4 or other DPP-related enzymes. In vitro data indicatethat CYP2D6 and CYP3A4 contribute to the limited metabolism of alogliptin.
Alogliptin exists predominantly as the (R)-enantiomer (more than 99%) and undergoes little or no chiral conversion in vivo to the (S)-enantiomer. The (S)-enantiomer is not detectable at the 25 mg dose.
Excretion: The primary route of elimination of [14C] alogliptin-derived radioactivity occurs via renal excretion (76%) with 13% recovered in the feces, achieving a total recovery of 89% of the administered radioactive dose. The renal clearance of alogliptin (9.6 L/hr) indicates some active renal tubular secretion and systemic clearance was 14.0 L/hr.
Special Populations: Renal Impairment: A single-dose, open-label study was conducted to evaluate the pharmacokinetics of alogliptin 50 mg in patients with chronic renal impairment compared with healthy subjects.
In patients with mild renal impairment (creatinine clearance [CrCl] ≥60 to <90 mL/min), an approximate 1.2-fold increase in plasma AUC of alogliptin was observed. Because increases of this magnitude are not considered clinically relevant, dose adjustment for patients with mild renal impairment is not recommended.
In patients with moderate renal impairment (CrCl ≥30 to <60 mL/min), an approximate two-fold increase in plasma AUC of alogliptin was observed. To maintain similar systemic exposures of NESINA to those with normal renal function, the recommended dose is 12.5 mg once daily in patients with moderate renal impairment.
In patients with severe renal impairment (CrCl ≥15 to <30 mL/min) and end-stage renal disease (ESRD) (CrCl <15 mL/min or requiring dialysis), an approximate three- and four-fold increase in plasma AUC of alogliptin were observed, respectively. Dialysis removed approximately 7% of the drug during a three-hour dialysis session. NESINA may be administered without regard to the timing of the dialysis. To maintain similar systemic exposures of NESINA to those with normal renal function, the recommended dose is 6.25 mg once daily in patients with severe renal impairment, as well as in patients with ESRD requiring dialysis.
Hepatic Impairment: Total exposure to alogliptin was approximately 10% lower and peak exposure was approximately 8% lower in patients with moderate hepatic impairment (Child-Pugh Grade B) compared to healthy subjects. The magnitude of these reductions is not considered to be clinically meaningful. Pharmacokinetics in patients with severe hepatic impairment (Child-Pugh Grade C) have not been studied. Use caution when administering NESINA to patients with liver disease (see Precautions).
Gender: No dose adjustment of NESINA is necessary based on gender. Gender did not have any clinically meaningful effect on the pharmacokinetics of alogliptin.
Geriatric: No dose adjustment of NESINA is necessary based on age. Age did not have any clinically meaningful effect on the pharmacokinetics of alogliptin.
Pediatric: Studies characterizing the pharmacokinetics of alogliptin in pediatric patients have not been performed.
Race: No dose adjustment of NESINA is necessary based on race. Race (White, Black, and Asian) did not have any clinically meaningful effect on the pharmacokinetics of alogliptin.
Drug Interactions: In Vitro Assessment of Drug Interactions: In vitro studies indicate that alogliptin is neither an inducer of CYP1A2, CYP2B6, CYP2C9, CYP2C19 and CYP3A4, nor an inhibitor of CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP3A4 and CYP2D6 at clinically relevant concentrations.
In Vivo Assessment of Drug Interactions: Effects of Alogliptin on the Pharmacokinetics of Other Drugs: In clinical studies, alogliptin did not meaningfully increase the systemic exposure to the following drugs that are metabolized by CYP isozymes or excreted unchanged in urine (Figure 3). No dose adjustment of NESINA is recommended based on results of the described pharmacokinetic studies. (See Figure 3.)
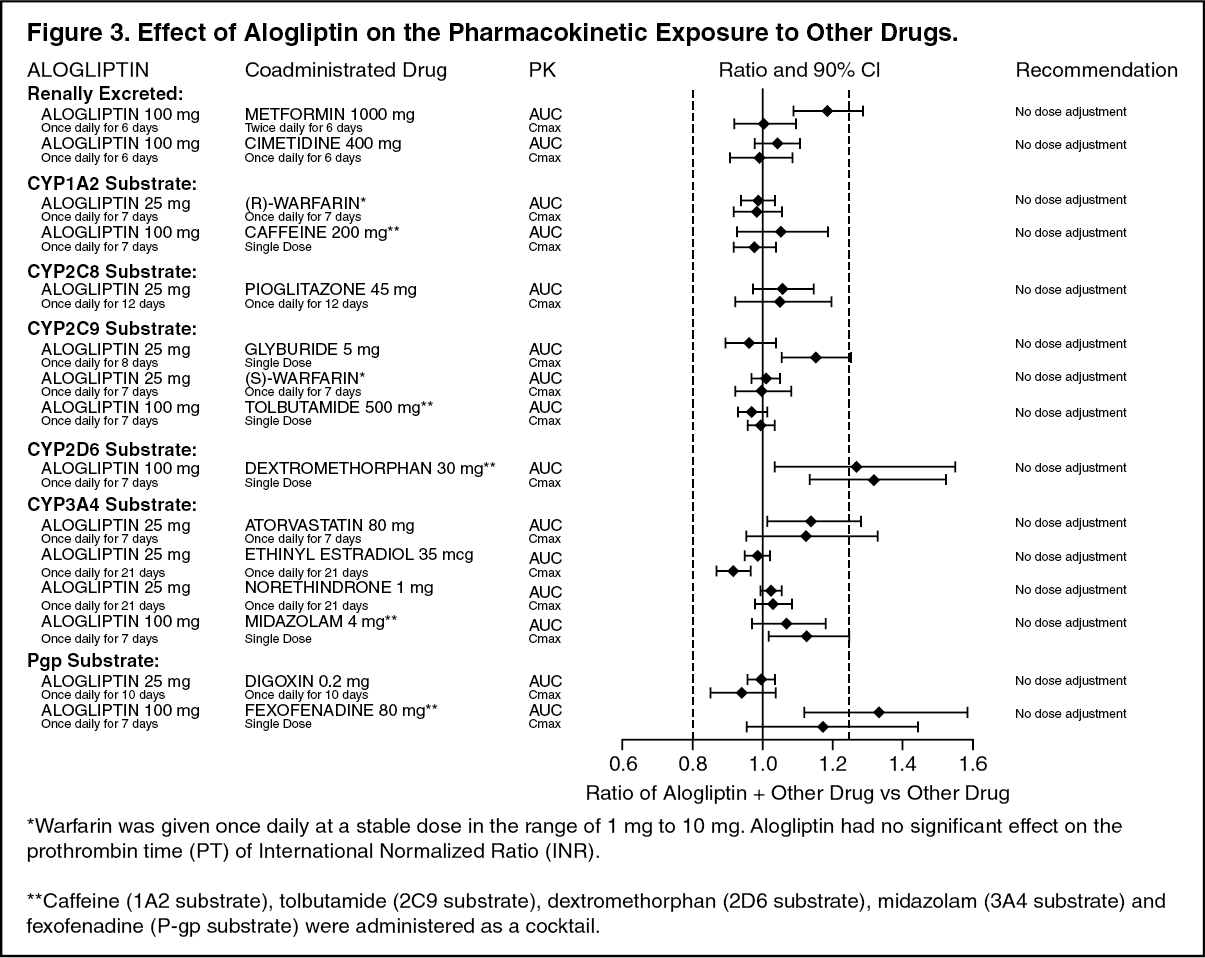 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageEffects of Other Drugs on the Pharmacokinetics of Alogliptin: There are no clinically meaningful changes in the pharmacokinetics of alogliptin when NESINA is administered concomitantly with the drugs described in Figure 4. (See Figure 4.)
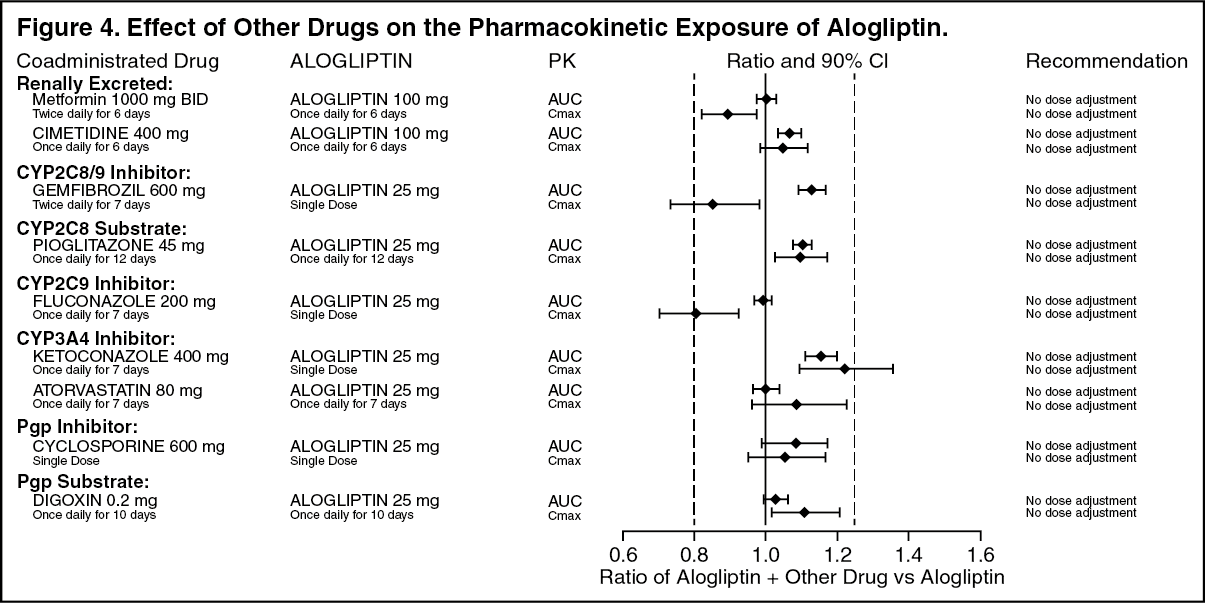 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageToxicology: Carcinogenesis, Mutagenesis, Impairment of Fertility: Rats were administered oral doses of 75, 400 and 800 mg/kg alogliptin for two years. No drug-related tumors were observed up to 75 mg/kg or approximately 32 times the maximum recommended clinical dose of 25 mg, based on area under the plasma concentration curve (AUC) exposure. At higher doses (approximately 308 times the maximum recommended clinical dose of 25 mg), a combination of thyroid C-cell adenomas and carcinomas increased in male but not female rats. No drug-related tumors were observed in mice after administration of 50, 150 or 300 mg/kg alogliptin for 2 years, or up to approximately 51-times the maximum recommended clinical dose of 25 mg, based on AUC exposure.
Alogliptin was not mutagenic or clastogenic, with and without metabolic activation, in the Ames test with S. typhimurium and E. coli or the cytogenetic assay in mouse lymphoma cells. Alogliptin was negative in the in vivo mouse micronucleus study.
In a fertility study in rats, alogliptin had no adverse effects on early embryonic development, mating or fertility at doses up to 500 mg/kg, or approximately 172-times the clinical dose based on plasma drug exposure (AUC).