Chemotherapy induced neutropenia: The recommended dose of NEUTROMAX is 0.5 M I.U. (5 µg)/kg body-weight (corresponding to 16.6 µL of injection solution) once daily. A single vial of NEUTROMAX of 30 M.I.U. therefore provides the daily dose required by a patient of 60 kg body-weight.
In patients who received myeloablative-chemotherapy followed by bone marrow transplantation: The recommended starting dose of NEUTROMAX is 1.0 M I.U. (10 µg)/kg/day. Given as a 30 minutes or 24 hours intravenous infusion or 1.0 M I.U. (10 µg)/kg/day given by continuous 24 hours subcutaneous infusion. NEUTROMAX should be diluted in 20 mL of 5% glucose solution.
Once the neutrophil nadir has been passed, the daily dose of NEUTROMAX should be adjusted as follows: See Table 2.
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
NEUTROMAX has been shown to be effective and well tolerated in this setting at doses up to 70 µg/kg/day.
Peripheral blood progenitor cell mobilization (PBPC) in autologous or allogeneic stem cell transplantation: The recommended dose of NEUTROMAX for PBPC mobilization is 1.0 M I.U. (10 µg)/kg/day subcutaneous injection or subcutaneous infusion once daily for 6-7 days with leukaphereses performed on days 5, 6 and 7.
NEUTROMAX should be maintained until the last leukapheresis. Leukapheresis should be continued until the cumulative CD34+ cell yield in the leukapheresis harvests is ≥5 x 10
6 cells/kg.
Severe chronic neutropenia (SCN): The recommended starting dose for congenital neutropenia is 1.2 M I.U. (12 µg)/kg/day subcutaneous infection (single or divided doses). The recommended starting dose for idiopathic or cyclic neutropenia is 0.5 M I.U. (5 µg)/kg/day subcutaneous injection (single or divided doses). Chronic administration is required to maintain adequate ANC. Dose should be doubled or halved after 1-2 weeks depending on counts. The dose of NEUTROMAX may be adjusted every 1 to 2 weeks to maintain ANC between 2 to 10 x 10
9/L. Doses may need reducing if vasculitis occurs.
Acute myelogenous leukemia (AML): NEUTROMAX use as an adjunct to acute myelogenous leukemia induction and consolidation therapy: The recommended dose of NEUTROMAX is 0.5 M I.U. (5 µg)/kg/day. It should be started at least 24 hours after chemotherapy and be discontinued at least 24 hours before the next dose of chemotherapy.
Further information: Clinical trials with NEUTROMAX have included a small number of elderly patients but special studies have not been performed in this group and therefore, specific dosage recommendations cannot be made.
The safety and efficacy of NEUTROMAX have not been established in children.
Administration: NEUTROMAX may be administered as a subcutaneous injection or as a short intravenous infusion, diluted in 5% glucose solution, given over 30 minutes (see "Instructions for Dilution" as follows). The first dose of NEUTROMAX should not be administered during the 24 hours following cytotoxic chemotherapy but within 24 hours of bone marrow infusion. Daily dosing with NEUTROMAX should continue until the expected neutrophil nadir is passed and the neutrophil count has recovered to the normal range. The required duration of treatment can be up to 14 days, depending on the type, dose and schedule of cytotoxic chemotherapy used. In patients receiving cytotoxic chemotherapy, a transient increase in neutrophil counts is typically seen 1 to 2 days after the beginning of NEUTROMAX therapy. However, for a sustained therapeutic response, NEUTROMAX therapy should not be discontinued before the expected nadir has passed and the neutrophil count has recovered to the normal range. Premature discontinuation of NEUTROMAX therapy, prior to the time of the expected neutrophil nadir, is not recommended.
In patients treated with cytotoxic chemotherapy and autologous bone marrow transplantation, NEUTROMAX may be given as a subcutaneous or intravenous infusion diluted in 20-50 mL 5% glucose solution (see "Instructions for Dilution" as follows and also "Dosage.").
The first dose of NEUTROMAX should not be administered before 24 hours following cytotoxic chemotherapy but should be within 24 hours of bone marrow infusion. The efficacy and safety of NEUTROMAX given for periods exceeding 28 days in this setting and group of patients have not been established.
Instructions for Dilution: If required, NEUTROMAX may be diluted in a 5% glucose solution. Diluted NEUTROMAX may be adsorbed to glass and plastic materials. However, when diluted according to the following instructions, NEUTROMAX is compatible with glass and a variety of plastics including PVC, polyolefin (a co-polymer of polypropylene and polyethylene) and polypropylene.
If NEUTROMAX is diluted to a concentration below 1.5 M 1.U. (15 µg) per mL, human serum albumin (HSA) should be added to a final concentration of 2 mg/mL.
Example: In a final injection volume of 20 mL, total doses of NEUTROMAX less than 30 M I.U. (300 µg) should be given with 0.2 mL of 20% human albumin solution (Ph.Eur.) added.
Dilution to a final concentration less than 0.2 M I.U. (2 µg) per mL is not recommended at any time.


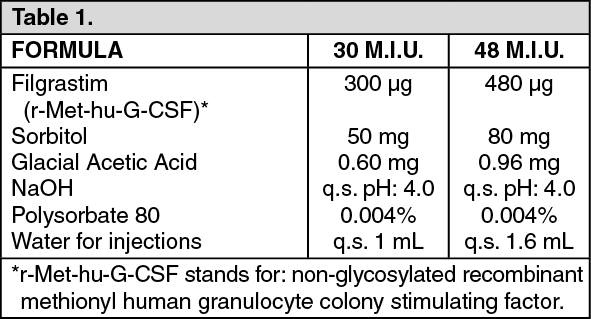 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image


 Sign Out
Sign Out