


 Sign Out
Sign Out

Pharmacology: Pharmacodynamics: Mechanism of action: Enfortumab vedotin is an ADC targeting Nectin-4, an adhesion protein located on the surface of the urothelial cancer cells. It is comprised of a fully human IgG1-kappa antibody conjugated to the microtubule-disrupting agent, MMAE, via a protease-cleavable linker. Nonclinical data suggest that the anticancer activity of enfortumab vedotin is due to the binding of the ADC to Nectin-4-expressing cells, followed by internalization of the ADC-Nectin-4 complex, and the release of MMAE via proteolytic cleavage. Release of MMAE disrupts the microtubule network within the cell, subsequently inducing cell cycle arrest and apoptosis, and immunogenic cell death. MMAE released from enfortumab vedotin targeted cells can diffuse into nearby Nectin-4 low-expressing cells resulting in cytotoxic cell death. Combination of enfortumab vedotin with PD-1 inhibitors results in enhanced anti-tumor activity, consistent with the complementary mechanisms of MMAE induced cell cytotoxicity and induction of immunogenic cell death, plus the up-regulation of immune function by PD-1 inhibition.
Pharmacodynamic effects: In an exposure-response analysis, a higher exposure was associated with higher incidence of some adverse reactions (e.g., Grade ≥2 peripheral neuropathy, Grade ≥3 hyperglycemia).
Cardiac Electrophysiology: The effect of Padcev on the duration of cardiac ventricular repolarization was evaluated in 17 patients with locally advanced or metastatic urothelial carcinoma who received Padcev on Days 1, 8, and 15 of each 28-day cycle. Based on concentration - QTcF modeling, a population mean change in QTcF interval (change from baseline QTcF; upper 1-sided 95% CI) of 6.17 (10.5) msec was estimated to occur at a geometric mean Cmax of 20.1 μ/mL for the ADC. For MMAE, a population mean change in QTcF interval (upper 1-sided 95% CI) of -3.14 (9.52) msec was estimated to occur at a geometric mean Cmax of 3.94 ng/mL. At the recommended dose of 1.25 mg/kg, Padcev had no large effect on QTc prolongation (>20 msec).
Clinical efficacy and safety: Urothelial Cancer: Previously Untreated Cisplatin Ineligible Patients with Locally Advanced or Metastatic Urothelial Carcinoma: EV-103: The efficacy of Padcev in combination with pembrolizumab was evaluated in a phase 2, open-label, multi-cohort (dose escalation cohort, Cohort A, Cohort K) study in patients with locally advanced or metastatic urothelial cancer who were ineligible for cisplatin-containing chemotherapy and received no prior systemic therapy for locally advanced or metastatic disease.
Patients in the dose escalation cohort (n=5) and Cohort A (n=40) received Padcev 1.25 mg/kg in combination with pembrolizumab 200 mg. Patients in Cohort K received Padcev 1.25 mg/kg as a single agent (n=73) or in combination with pembrolizumab 200 mg (n=76).
Patients received Padcev 1.25 mg/kg as an intravenous infusion over 30 minutes on Days 1 and 8 of a 21-day cycle followed by pembrolizumab 200 mg on Day 1 of a 21-day cycle approximately 30 minutes after Padcev until disease progression or unacceptable toxicity.
Reasons for cisplatin ineligibility in patients enrolled in EV-103 included: ECOG PS of 2, creatinine clearance ≥30 and <60 mL/min, hearing loss/dysfunction and/or age.
Patients with active CNS metastases, ongoing sensory or motor neuropathy Grade ≥2, or uncontrolled diabetes defined as HbA1c ≥8% or HbA1c ≥7% with associated diabetes symptoms were excluded from participating in the study.
A total of 121 patients received Padcev 1.25 mg/kg in combination with pembrolizumab. The median age was 71 years (range: 51 to 91); 74% were male; 85% were White; and 45% of patients had an ECOG performance status of 1 and 15% had an ECOG performance status of 2. Forty-seven percent of patients had a documented baseline HbA1c of <5.7%. At baseline, 98% of patients had metastatic urothelial cancer and 2.5% of patients had locally advanced urothelial cancer. Eighty four percent of patients had visceral metastasis at baseline including 22% with liver metastases. Of the 108 patients tested who had tissue evaluable for PD-L1 expression, 43% of patients had tumors that expressed PD-L1 with a CPS ≥10 and 57% had tumors that expressed PD-L1 with a CPS <10. The median follow-up time for the dose escalation cohort + Cohort A was 44.7 months (range: 0.7 to 52.4) and for Cohort K was 14.8 months (range: 0.6 to 26.2).
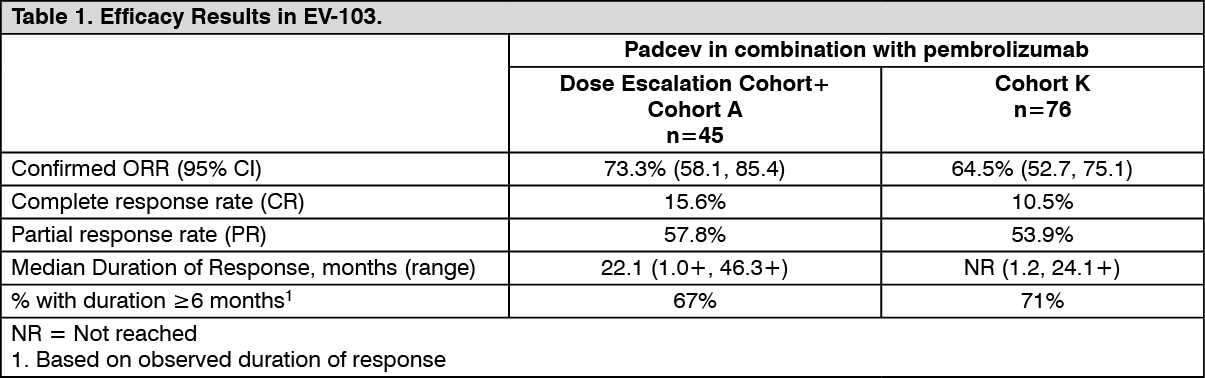
Confirmed ORR was evaluated by BICR using RECIST v1.1. The median time to response was 1.94 months (range: 1.1 to 13.2) for the dose escalation cohort + Cohort A and was 2.07 months (range: 1.1 to 6.6) for Cohort K. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn the combined efficacy analysis of the dose escalation cohort, Cohort A and Cohort K, (n=121), confirmed ORR was 68% (95% CI: 58.7, 76.0) with complete and partial response rates of 12% and 55%, respectively. Among the responding patients, 80% had responses of 6 months or longer (based on observed duration of response).
Previously Treated Patients with Locally Advanced or Metastatic Urothelial Carcinoma: EV-301: The efficacy of Padcev as a single agent was evaluated in EV-301, an open-label, randomized, phase 3, multicenter study that enrolled 608 patients with locally advanced or metastatic urothelial cancer who received prior treatment with a PD-1 or PD-L1 inhibitor and a platinum-containing chemotherapy. Patients were randomized 1:1 to receive either Padcev 1.25 mg/kg on Days 1, 8 and 15 of a 28-day cycle or one of the following chemotherapies as decided by the investigator: docetaxel (38%), paclitaxel (36%) or vinflunine (26%).
Patients were excluded from the study if they had active CNS metastases, ongoing sensory or motor neuropathy Grade ≥2, or uncontrolled diabetes defined as hemoglobin A1C (HbA1c) ≥8% or HbA1c ≥7% with associated diabetes symptoms.
The median age was 68 years (range: 30 to 88 years), 77% were male, and most patients were White (52%) or Asian (33%). All patients had a baseline Eastern Cooperative Oncology Group (ECOG) performance status of 0 (40%) or 1 (60%). Eighty percent of patients had visceral metastases including 31% with liver metastases. Seventy-six percent of patients had urothelial carcinoma/transitional cell carcinoma (TCC) histology and 14% had urothelial carcinoma mixed. A total of 527 out of 608 subjects had evaluable Nectin-4 results; of these 527 subjects, 516 (98%) had detectable Nectin-4 (H-score >0) as assessed by a validated immunohistochemistry (IHC) assay. A total of 76 (13%) of patients received ≥3 lines of prior systemic therapy. Fifty-two percent (314) of patients received prior PD-1 inhibitor, 47% (284) received prior PD-L1 inhibitor, and an additional 1% (9) patients received both PD-1 and PD-L1 inhibitors. Sixty-nine percent of patients did not respond to prior therapy with a PD-1 or PD-L1 inhibitor. Sixty-three percent (383) of patients received prior cisplatin-based regimens, 26% (159) received prior carboplatin-based regimens, and an additional 11% (65) received both cisplatin and carboplatin-based regimens.
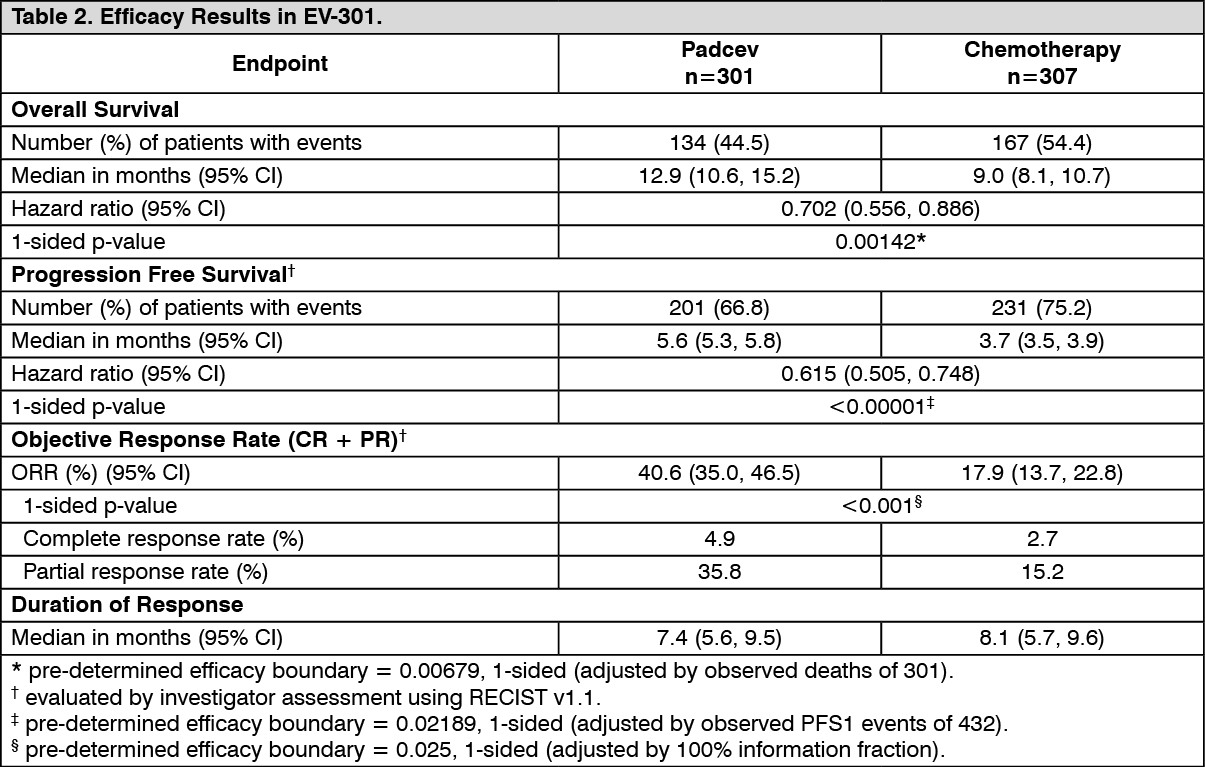
Table 2 summarizes the efficacy results for the EV 301 study, after a median follow up time of 11.1 months (95% CI: 10.6 to 11.6). (See Table 2 and figure.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image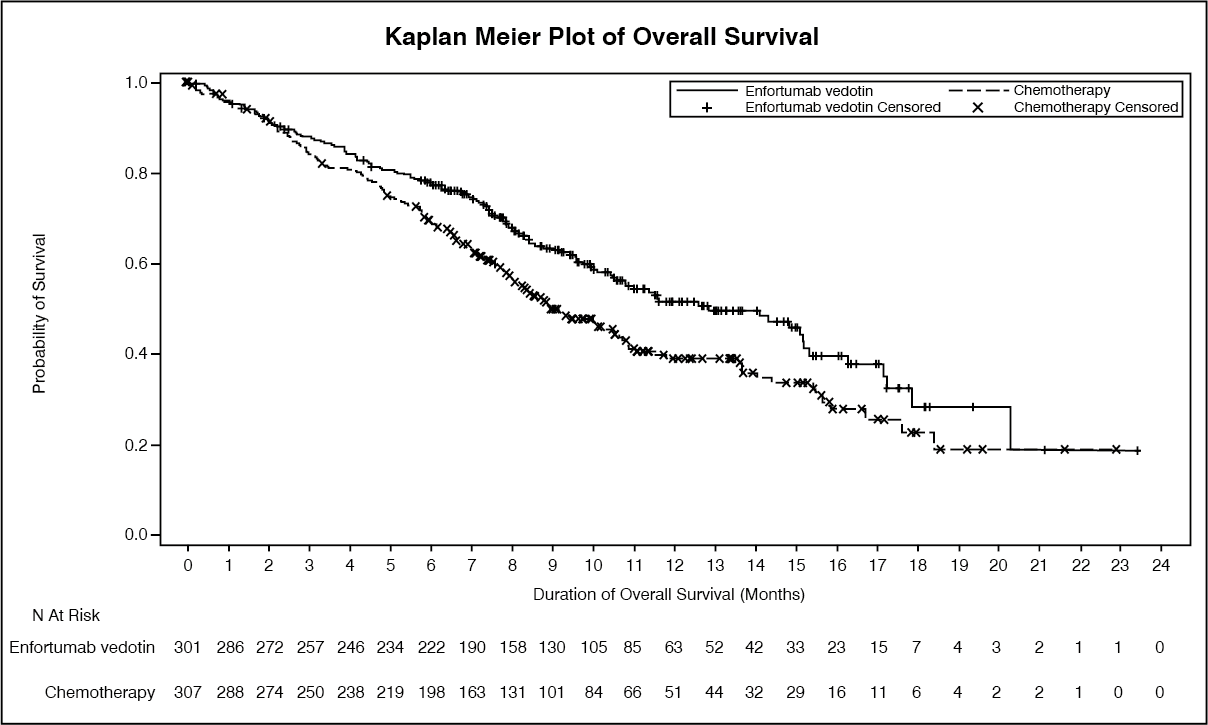 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePharmacokinetics: Population pharmacokinetic analysis included data from 748 patients treated with enfortumab vedotin as a single agent in clinical studies. Enfortumab vedotin pharmacokinetics were characterized after single and multiple doses in patients with locally advanced or metastatic urothelial carcinoma and other solid tumors.
The pharmacokinetics of ADC and unconjugated MMAE were consistent when assessed following enfortumab vedotin as a single agent and in combination with pembrolizumab.
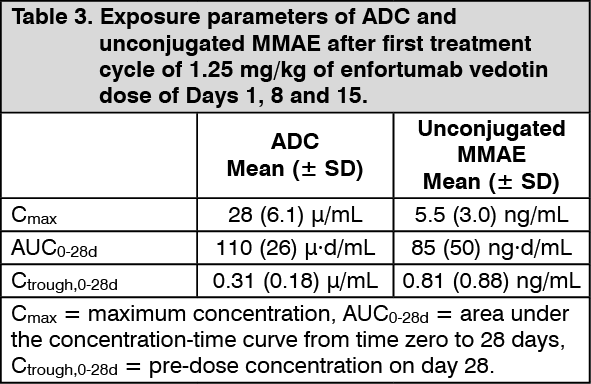
Peak ADC concentrations were observed near the end of intravenous infusion administration [median estimate of 0.03 days (~0.72 hours)] and peak unconjugated MMAE concentrations were observed approximately 2 days after enfortumab vedotin dosing. After repeat administration of enfortumab vedotin at 1.25 mg/kg on Days 1, 8 and 15 of a 28-day cycle or on Days 1 and 8 of a 21-day cycle, minimal to no accumulation of ADC or unconjugated MMAE was observed. ADC and unconjugated MMAE concentrations appeared to reach steady state after 1 cycle. (See Table 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageDistribution: The mean estimate of steady-state volume of distribution of ADC was 12.8 L following 1.25 mg/kg of enfortumab vedotin.
In vitro, the binding of unconjugated MMAE to human plasma proteins ranged from 68% to 82%. Unconjugated MMAE is not likely to displace or to be displaced by highly protein-bound drugs. In vitro studies indicate that unconjugated MMAE is a substrate of P-glycoprotein.
Biotransformation: A small fraction of unconjugated MMAE released from enfortumab vedotin is metabolized. In vitro data indicate that the metabolism of MMAE occurs primarily via oxidation by CYP3A4.
Elimination: The mean clearance (CL) of ADC and unconjugated MMAE in patients was 0.114 L/h and 2.11 L/h, respectively.
ADC elimination exhibited a multi-exponential decline with a half-life of 3.6 days.
Elimination of unconjugated MMAE appeared to be limited by its rate of release from enfortumab vedotin. Unconjugated MMAE elimination exhibited a multi-exponential decline with a half-life of 2.6 days.
Excretion: The excretion of unconjugated MMAE occurs mainly in feces with a smaller proportion in urine. After a single dose of another ADC that contained unconjugated MMAE, approximately 24% of the total unconjugated MMAE administered was recovered in feces and urine as unchanged unconjugated MMAE over a 1-week period. The majority of recovered unconjugated MMAE was excreted in feces (72%). A similar excretion profile is expected for unconjugated MMAE after enfortumab vedotin administration.
Special Populations: Elderly: Population pharmacokinetic analysis indicates that age [range: 24 to 90 years; 60% (450/748) >65 years, 19% (143/748) >75 years] does not have a clinically meaningful effect on the pharmacokinetics of enfortumab vedotin.
Race and gender: Based on population pharmacokinetic analysis, race [69% (519/748) White, 21% (158/748) Asian, 1% (10/748) Black and 8% (61/748) others or unknown] and gender [73% (544/748) male] do not have a clinically meaningful effect on the pharmacokinetics of enfortumab vedotin.
Renal impairment: The pharmacokinetics of ADC and unconjugated MMAE were evaluated after the administration of 1.25 mg/kg of enfortumab vedotin to patients with mild (CrCL >60-90 mL/min; n=272), moderate (CrCL 30-60 mL/min; n=315) and severe (CrCL 15-<30 mL/min; n=25) renal impairment. No significant differences in AUC exposure of ADC or unconjugated MMAE were observed in patients with mild, moderate or severe renal impairment compared to patients with normal renal function. Enfortumab vedotin has not been evaluated in patients with end stage renal disease (CrCL <15 mL/min).
Hepatic impairment: Based on population pharmacokinetics analysis using data from clinical studies in patients with metastatic UC, there was no significant differences in ADC exposure and a 37% increase in unconjugated MMAE AUC were observed in patients with mild hepatic impairment (total bilirubin 1 to 1.5 x ULN and AST any, or total bilirubin ≤ULN and AST >ULN, n=65) compared to patients with normal hepatic function. Enfortumab vedotin has only been studied in a limited number of patients with moderate hepatic impairment (n=3) and has not been evaluated in patients with severe hepatic impairment. The effect of moderate or severe hepatic impairment (total bilirubin >1.5 x ULN and AST any) or liver transplantation on the pharmacokinetics of ADC or unconjugated MMAE is unknown.
Drug-drug interactions: Formal drug-drug interaction studies with enfortumab vedotin have not been conducted. Physiologically-based pharmacokinetic modeling was conducted to predict the drug-drug interaction potential of unconjugated MMAE.
Effects of Other Drugs on Enfortumab Vedotin: Physiologically-Based Pharmacokinetic Modeling Predictions: Strong CYP3A Inhibitor: Concomitant use of enfortumab vedotin with ketoconazole (a combined P-gp and strong CYP3A inhibitor) is predicted to increase unconjugated MMAE Cmax by 15% and AUC by 38%, with no change in ADC exposure.
Strong CYP3A Inducer: Concomitant use of enfortumab vedotin with rifampin (a combined P-gp and strong CYP3A inducer) is predicted to decrease unconjugated MMAE Cmax by 28% and AUC by 53%, with no change in ADC exposure. The full impact of rifampin on the Cmax of MMAE may be underestimated in the PBPK model.
Effects of Enfortumab Vedotin on Other Drugs: Concomitant use of enfortumab vedotin is predicted not to affect exposure to midazolam (a sensitive CYP3A substrate) or digoxin (a P-gp substrate). In vitro studies using human liver microsomes indicate that unconjugated MMAE inhibits CYP3A4/5 but not other CYP450 isoforms. Unconjugated MMAE did not induce major CYP450 enzymes in human hepatocytes.
In vitro studies indicate that unconjugated MMAE is a substrate and not an inhibitor of the efflux transporter P-glycoprotein (P-gp). In vitro studies determined that unconjugated MMAE was not a substrate of breast cancer resistance protein (BCRP), multidrug resistance-associated protein 2 (MRP2), organic anion transporting polypeptide 1B1 or 1B3 (OATP1B1 or OATP1B3), organic cation transporter 2 (OCT2), or organic anion transporter 1 or 3 (OAT1 or OAT3). Unconjugated MMAE was not an inhibitor of the bile salt export pump (BSEP), P-gp, BCRP, MRP2, OCT1, OCT2, OAT1, OAT3, OATP1B1, or OATP1B3 at clinically relevant concentrations.
Toxicology: Preclinical safety data: Skin lesions were noted repeated dose studies in rats (4- and 13-weeks) and in monkeys (4-weeks). The skin changes were fully reversible by the end of a 6-week recovery period.
Hyperglycemia reported in the clinical studies was absent in both the rat and monkey toxicity studies and there were no histopathological findings in the pancreas of either species.
Genotoxicity studies showed that MMAE had no discernible genotoxic potential in a reverse mutation test in bacteria (Ames test) or in a L5178Y TK+/- mouse lymphoma mutation assay. MMAE did induce chromosomal aberrations in the micronucleus test in rats which is consistent with the pharmacological action of microtubule-disrupting agents.
In a rat embryo-fetal development toxicity study, enfortumab vedotin resulted in a dose-related (2 or 5 mg/kg) decrease in maternal body weight gain and reduced food consumption at the 5 mg/kg dose level. Clinical observations included fur loss at both dose levels (one animal per dose level) as well as scabbing of the skin on the back or ventral aspect in one animal at the 5 mg/kg level.
Fetal toxicity was noted at both the 2- and 5 mg/kg dose levels (1- and 3-fold the human Cmax, respectively) with reduced litter size noted at the 2 mg/kg dose level and complete litter loss in the 5 mg/kg/day dose group. The decrease in the litter size was reflected in an increase in early resorptions. Mean fetal body weight in the surviving fetuses at the 2 mg/kg dose level were reduced compared with control.
Enfortumab vedotin associated fetal skeletal variations were considered developmental delays. A dose of 2 mg/kg (approximately similar to the exposure at the recommended human dose) resulted in maternal toxicity, embryo-fetal lethality and structural malformations that included gastroschisis, malrotated hindlimb, absent forepaw, malpositioned internal organs and fused cervical arch. Additionally, skeletal anomalies (asymmetric, fused, incompletely ossified, and misshapen sternebrae, misshapen cervical arch, and unilateral ossification of the thoracic centra) and decreased fetal weight were observed
In addition, intravenous administration of MMAE (0.2 mg/kg; Cmax 1.1-fold the human Cmax at the recommended clinical dose) on Gestation Day 6 and 13 resulted in embryo-fetal lethality and fetal external malformations (protruding tongue, malrotated hindlimbs, gastroschisis, and agnathia).
Testicular toxicity was noted only in rats. Findings included seminiferous tubule degeneration and hypospermia in the epididymis (≥2.0 mg/kg; approximately 1-fold the human systemic exposure at the clinically recommended dose). These findings were partially reversed by the end of a 24-week recovery period. Testicular toxicity was not observed in sexually immature male monkeys administered enfortumab vedotin at doses up to 6 mg/kg (6-fold the human systemic exposure at the clinically recommended dose).
While not observed with enfortumab vedotin, ovarian effects were observed in repeat dose toxicity studies of other MMAE-containing ADCs. A mild to moderate decrease in, or absence of, secondary and tertiary ovarian follicles was observed in young female cynomolgus monkeys at doses ≥3 mg/kg weekly for 4 weeks. No changes were observed in primordial follicles. Effects on the secondary and tertiary ovarian follicles showed evidence of recovery 6 weeks after the end of dosing.
No dedicated preclinical safety studies were conducted with enfortumab vedotin in combination with pembrolizumab.