


 Sign Out
Sign Out

PHARMACOLOGY: Pharmacodynamics: Mechanism of Action: Entrectinib is a potent inhibitor of receptor tyrosine kinases TRKA, TRKB and TRKC (encoded by the neurotrophic tyrosine receptor kinase [NTRK] genes NTRK1, NTRK2 and NTRK3, respectively), proto-oncogene tyrosine-protein kinase ROS (ROS1; encoded by the gene ROS1), and anaplastic lymphoma kinase (ALK; encoded by the gene ALK). The major active metabolite of entrectinib, M5, showed similar in vitro potency and activity.
Fusion proteins that include TRK, ROS1 or ALK kinase domains drive tumorigenic potential through hyperactivation of downstream signaling pathways leading to unconstrained cell proliferation. Entrectinib potently inhibits the TRK kinases, ROS1 and ALK, leading to inhibition of downstream signaling pathways, cell proliferation and induction of tumor cell apoptosis. Entrectinib demonstrates potent inhibition of cancer cell lines harboring NTRK, ROS1 and ALK fusion genes, irrespective of tumor type. Entrectinib has anti-tumor potency in NTRK and ROS1 fusion-driven tumor models, driving tumor regressions across multiple tumor types, including sarcomas, head and neck carcinoma, non-small cell lung carcinoma (NSCLC), colorectal cancer (CRC), acute myeloid leukemia (AML), and gliomas.
Entrectinib is a CNS penetrant molecule that showed brain-to-plasma concentration ratios of 0.4-2.2 in multiple animal species (mice, rats and dogs). It has demonstrated potent anti-tumor activity in three TRKA-driven intracranial tumor models and one ALK-driven intracranial tumor model. These data are consistent with entrectinib dosing resulting in sufficient brain exposure achieving target pharmacological activities at steady-state and at clinically relevant systemic exposures.
Clinical/Efficacy Studies: NTRK fusion-positive solid tumors: Efficacy in Adult patients: The efficacy of ROZLYTREK in the treatment of NTRK fusion-positive solid tumors in adult patients was evaluated by combining the results from 3 single-arm, open label clinical trials (ALKA, STARTRK-1 and STARTRK-2) through a pre-specified integrated analysis.
Study ALKA was a Phase I single arm, open-label study in patients ≥ 18 years of age with solid tumors with NTRK1/2/3, ROS1, or ALK molecular alterations to determine the maximum tolerated dose. Study STARTRK-1 was a Phase I multi-center single arm, open label study in patients ≥ 18 years of age with solid tumors with NTRK1/2/3, ROS1, or ALK molecular alterations. The study included a dose escalation segment and a dose expansion segment. In the dose expansion segment, patients received 600 mg daily in repeated 4-week cycles and the primary objective was to evaluate the recommended Phase 2 dose. Study STARTRK-2 was a multicenter, international Phase II single-arm basket study in patients with solid tumors with NTRK1/2/3, ROS1, or ALK gene rearrangements. Patients received 600 mg ROZLYTREK once daily in 4-week cycles.
The primary efficacy outcome measures in the integrated analyses were objective response rate (ORR) and duration of response (DOR) as evaluated by Blinded Independent Central Review (BICR) according to Response Evaluation Criteria in Solid Tumors (RECIST) v1.1. The secondary efficacy outcome measures included clinical benefit rate (CBR), progression-free survival (PFS), time to central nervous system (CNS) progression, overall survival (OS), and in patients presenting with CNS metastases at baseline - intracranial (IC) ORR, IC-DOR, and IC-PFS (also evaluated by BICR using RECIST v1.1).
The efficacy evaluable analyses set comprised a total of 74 adult patients with confirmed NTRK fusion-positive solid tumors treated with ROZLYTREK, not previously treated with a TRK inhibitor, presenting with measurable disease at baseline as assessed by investigator, and with ≥ 6 months of follow up. NTRK fusion-positive status was determined by a validated nucleic acid-based test performed at a Clinical Laboratory Improvement Amendments (CLIA)-certified or equivalently accredited laboratory, prior to enrollment in the study.
The baseline demographic and disease characteristics of the efficacy evaluable population were: 47.3% males, median age of 57 years (range: 21 to 83 years), 70.0% white Caucasian, 17.6% Asian, 5.5% Hispanic or Latino and 59.7% never smokers. The ECOG (Eastern Cooperative Oncology Group) performance status at baseline was 0 (40.5%), 1 (45.9%), or 2 (13.5%). Most patients (97.3%) had metastatic disease [most common sites being lung (60.8%), lymph nodes (52.7%) and brain (25.7%)], 2.7% patients had locally advanced disease, and 27% patients had no prior systemic therapies. The overall median duration of follow-up was 14.2 months.
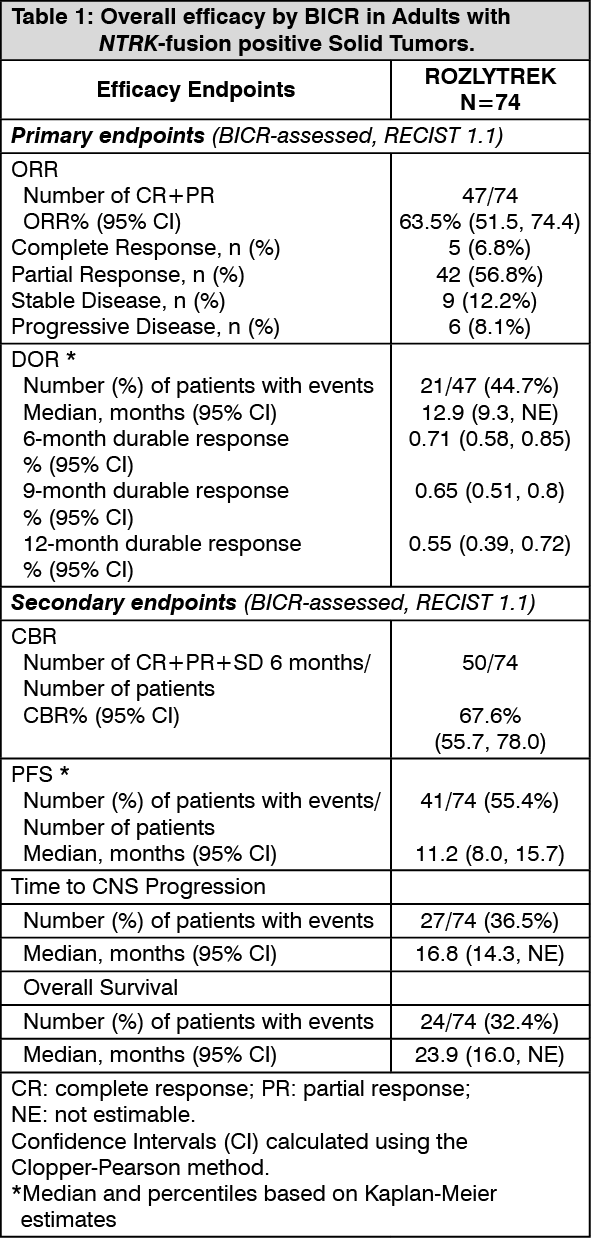
Efficacy results from patients with NTRK-fusion positive solid tumors are summarized in Table 1. (See Table 1.)
 Click on icon to see table/diagram/image
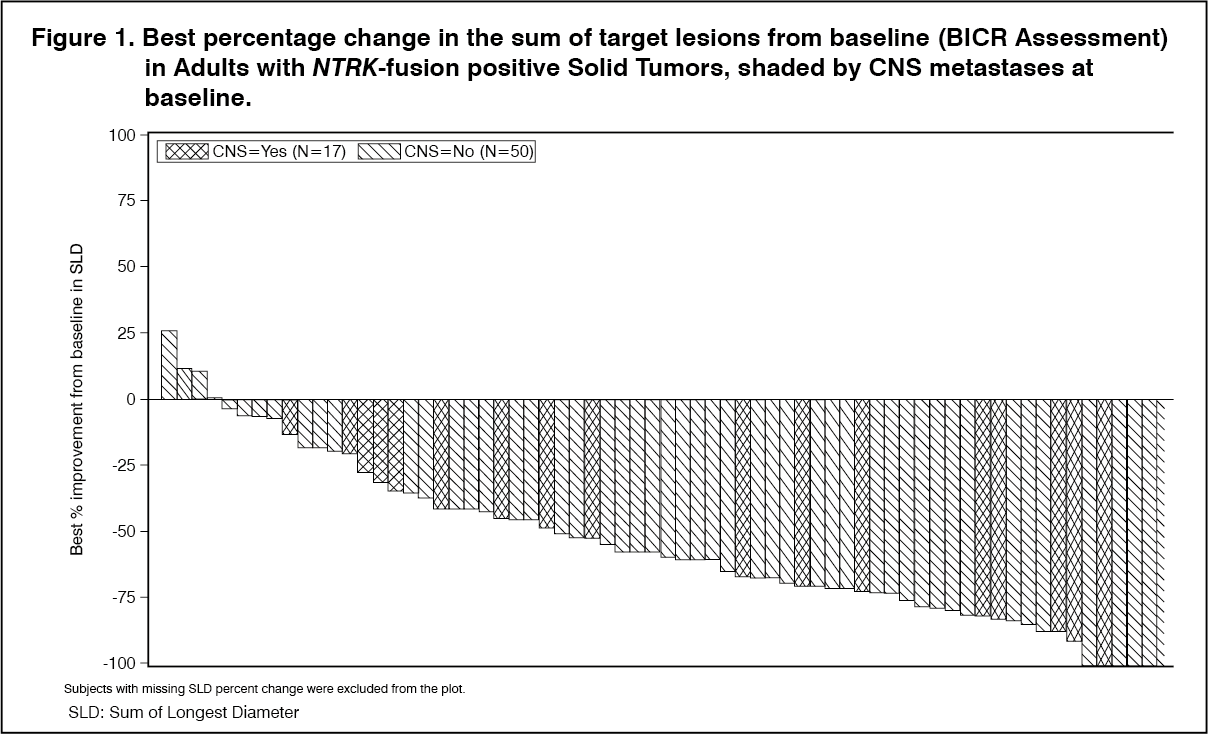
Click on icon to see table/diagram/imageAs shown in Figure 1, most adult patients with NTRK-fusion positive solid tumors experienced tumor shrinkage, as assessed by BICR according to RECIST 1.1. (See Figure 1.)
 Click on icon to see table/diagram/image
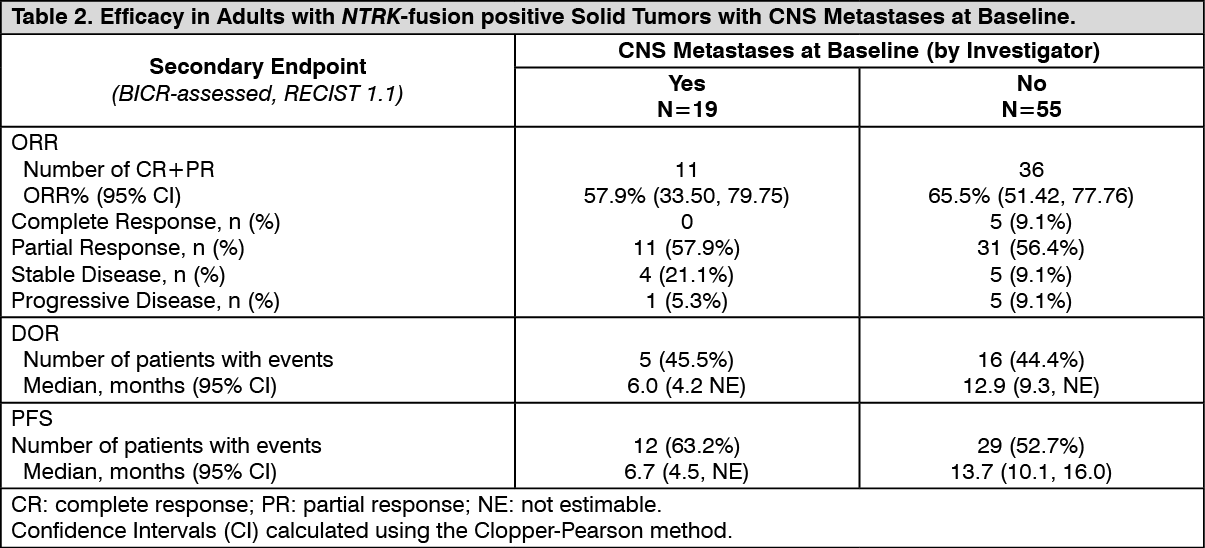
Click on icon to see table/diagram/imageOf the 74 adult patients with NTRK-fusion positive solid tumors in the efficacy evaluable analysis set, 19 patients were identified by the Investigator to have CNS metastases at baseline. Efficacy results by BICR according to RECIST v1.1 in this subgroup of patients with CNS metastases at baseline are summarized in Table 2. (See Table 2.)
 Click on icon to see table/diagram/image
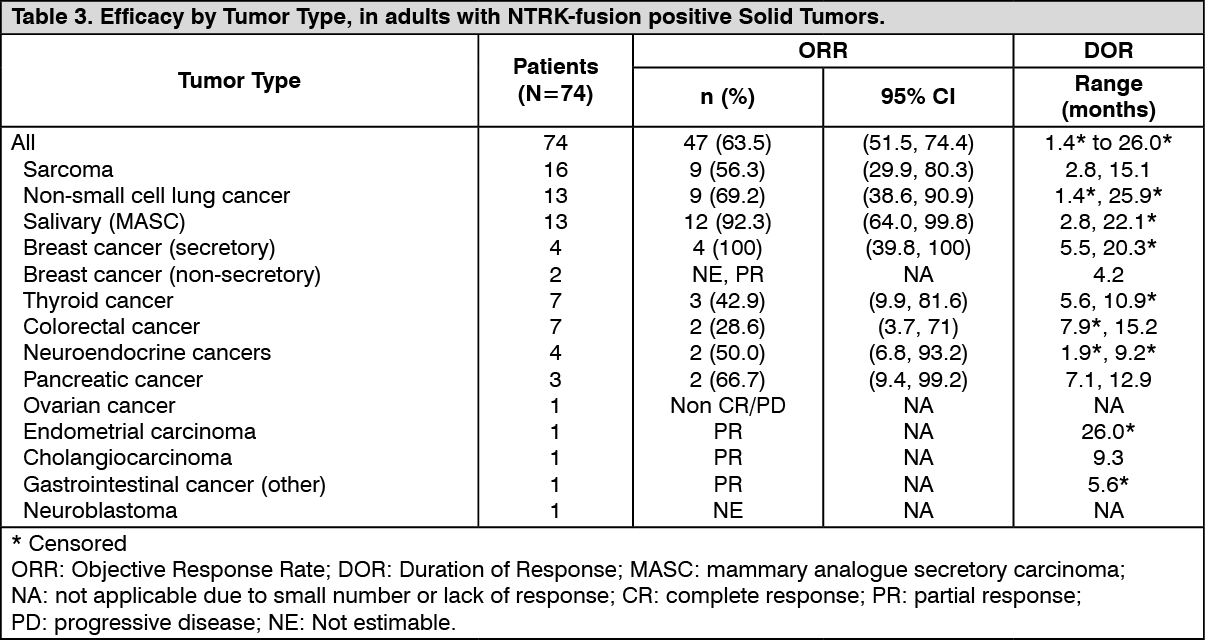
Click on icon to see table/diagram/imageObjective response rate by tumor type in all efficacy evaluable adult patients with NTRK-fusion positive solid tumors is presented in Table 3. (See Table 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIntracranial Response: Of the 74 adult patients with NTRK-fusion positive solid tumors in the efficacy evaluable analysis set, 16 patients had CNS metastases at baseline as assessed by BICR, including 8 patients with measurable CNS lesions. Intracranial ORR, DOR, and PFS assessed by BICR according to RECIST version 1.1 in this subgroup of patients with measurable CNS lesions at baseline are summarized in Table 4. (See Table 4.)
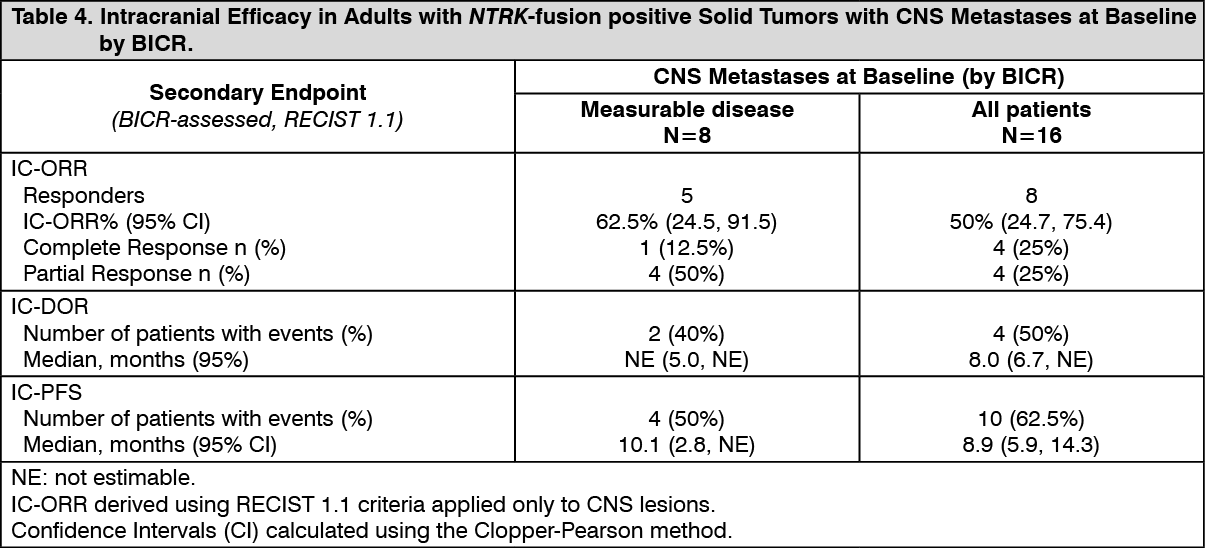 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePrimary CNS tumors: Across the three trials, seven adult patients with CNS primary tumors were treated with ROZLYTREK with a minimum of 6 months of follow-up. IC-ORR, DOR and PFS were assessed by BICR according to Response Assessment in Neuro-Oncology Criteria (RANO). One patient had an objective response with a DOR of 2.79 months and PFS of 6.34 months.
Patient Reported Outcomes: Study STARTRK-2 evaluated patient-reported outcomes (PRO) of the treatment impact on symptoms, functioning and health-related quality of life (HRQoL) based on the EORTC Core Quality of Life Questionnaire (QLQ-C30), Lung Cancer Module (QLQ-LC13), and the Colorectal Cancer Module (QLQ-CR29).
Most safety evaluable patients indicated that the symptoms commonly associated with ROZLYTREK treatment (lack of appetite, nausea, diarrhea and vomiting) were of low severity, if present. Efficacy evaluable patients with NTRK-fusion positive NSCLC (n=12) reported moderate lung-related symptoms at baseline with qualitative trends towards improvement while receiving Rozlytrek. Patients with mCRC (n=7) reported low abdominal symptoms burden at baseline, which generally remained low over time. Qualitative trends towards improvements in functioning (moderate to high at baseline) and maintenance of HRQoL (high at baseline) were observed for patients with NTRK fusion-positive solid tumors while receiving ROZLYTREK as measured by the Physical function, the Role function and the Global Health Status from the EORTC QLQ-C30.
Efficacy in Pediatric patients: The efficacy of ROZLYTREK in pediatric patients with NTRK fusion-positive solid tumors was evaluated in study STARTRK-NG (ST-NG). This study is a multi-center Phase I/II open-label dose-escalation and expansion study in pediatric patients with relapsed or refractory solid tumors, including primary CNS tumors, with or without NTRK, ROS1 or ALK molecular alterations. Patients received 250 mg/m2 to 750 mg/m2 once daily of ROZLYTREK in 4-week cycles. The range of survival follow up was 6.9 months to 18.0 months.
Table 5 summarizes the efficacy of ROZLYTREK in 7 pediatric patients (less than 18 years of age) with NTRK fusion-positive solid tumors as assessed by the Investigator according to RECIST version 1.1 for extra-cranial tumors and according to RANO for CNS primary tumors. Efficacy data in pediatric patients with NTRK fusion-positive solid tumors is further supported by extrapolation from results in the respective adult populations. (See Table 5.)
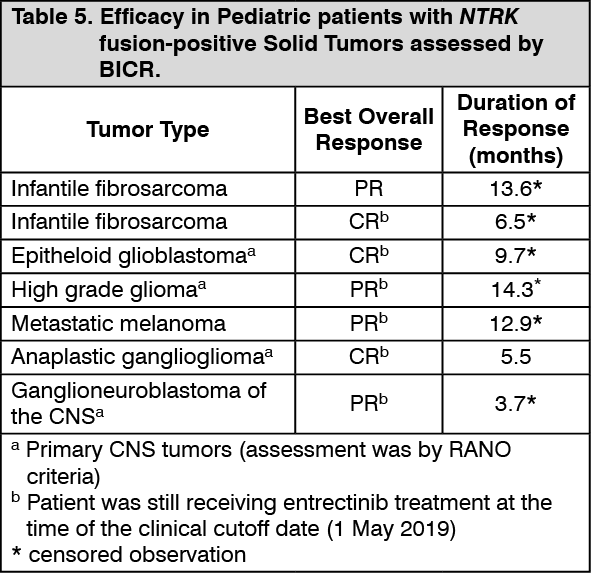 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageROS1-positive NSCLC: The efficacy of ROZLYTREK in the treatment of ROS1 positive locally advanced or metastatic NSCLC was evaluated by combining the results from 3 single-arm, open label clinical trials (ALKA, STARTRK-1 and STARTRK-2) described previously, through a pre-specified integrated analysis.
The primary efficacy outcome measures in the integrated analyses were ORR and DOR, as evaluated by BICR according to RECIST v1.1. The secondary efficacy outcome measures included CBR, PFS, time to CNS progression, OS, and in patients presenting with CNS metastases at baseline - IC-ORR, IC-DOR, and IC-PFS (also evaluated by BICR using RECIST v1.1).
The efficacy evaluable analyses set comprised a total of 94 patients with histologically confirmed ROS1-positive NSCLC treated with ROZLYTREK, not previously treated with a ROS1-inhibitor, presenting with measurable disease at baseline as assessed by the investigator, and with ≥12 months of follow up. ROS1-positive status was determined by a validated nucleic acid-based test performed at a CLIA-certified or equivalently accredited laboratory, prior to enrollment in the study.
The baseline demographic and disease characteristics of the efficacy evaluable population were: 36.2% males, median age of 53 years (range: 27 to 86 years), 79.8% patients <65 years of age, 48.9% white Caucasian, 43.6% Asian, 5.3% Black, 2.4% Hispanic or Latino and 59.6% never smokers. The ECOG (Eastern Cooperative Oncology Group) performance status at baseline was 0 (37.2%), 1 (51.1%), or 2 (11.7%). Most patients (98.9%) had metastatic disease with 42.6% having brain metastases [other common sites were lung (57.4%) and lymph nodes (75.5%)], 1.1% patients had locally advanced disease, and 33% patients had no prior systemic therapies. The overall median duration of follow-up was 20.9 months.
Efficacy results from patients with ROS1-positive NSCLC are summarized in Table 6. (See Table 6.)
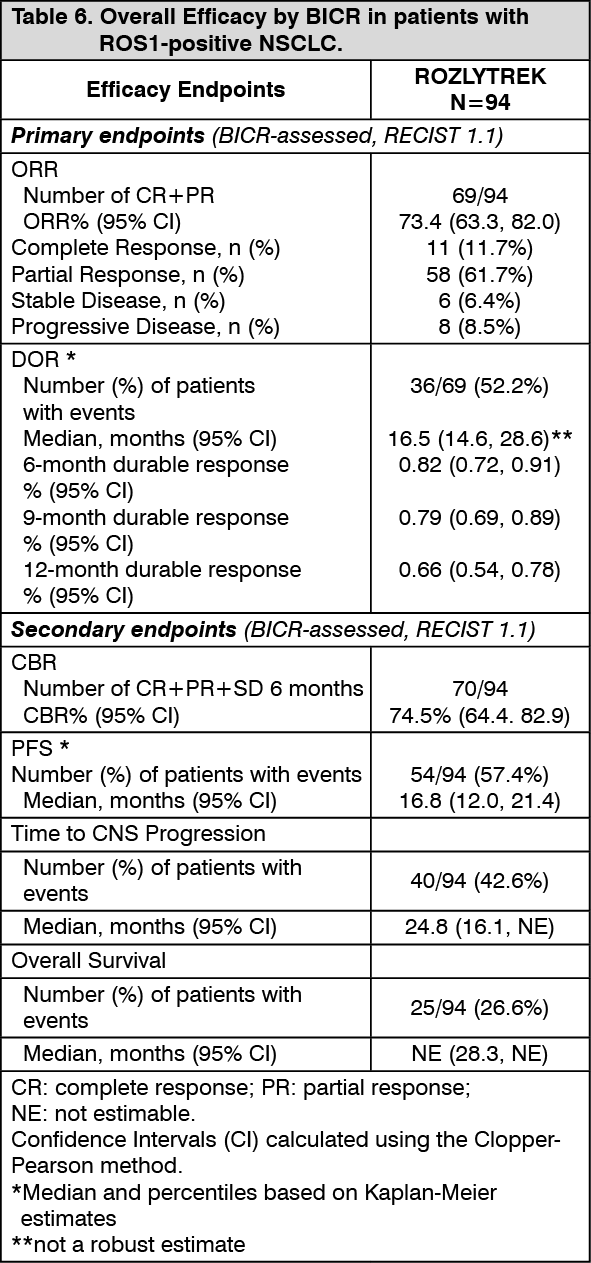 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageMost ROS1-positive NSCLC patients treated with ROZLYTREK experienced tumor shrinkage of their defined target lesions, as assessed by BICR according to RECIST 1.1. (See Figure 2.)
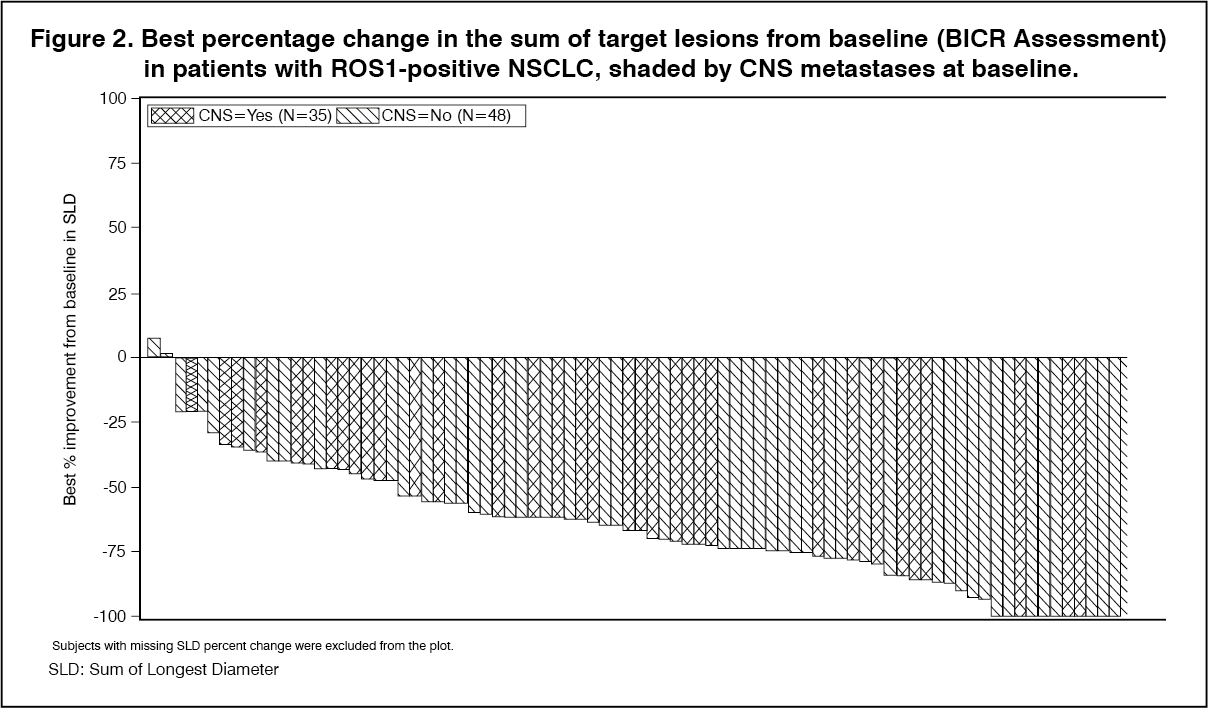 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageOf the 94 patients with ROS1-positive NSCLC in the efficacy evaluable analysis set, 40 patients were identified by the Investigator to have CNS metastases at baseline. Efficacy results by BICR according to RECIST v1.1 in this subgroup of patients with CNS metastases at baseline are summarized in Table 7. (See Table 7.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIntracranial Response: Of the 94 patients with ROS-1 positive NSCLC in the efficacy evaluable analysis set, 34 patients had CNS metastases at baseline as assessed by BICR, including 18 patients with measurable CNS lesions. Intracranial ORR, DOR, and PFS assessed by BICR according to RECIST version 1.1 in this subgroup of patients with measurable CNS lesions at baseline are summarized in Table 8 and Figure 3 as follows. (See Table 8 and Figure 3.)
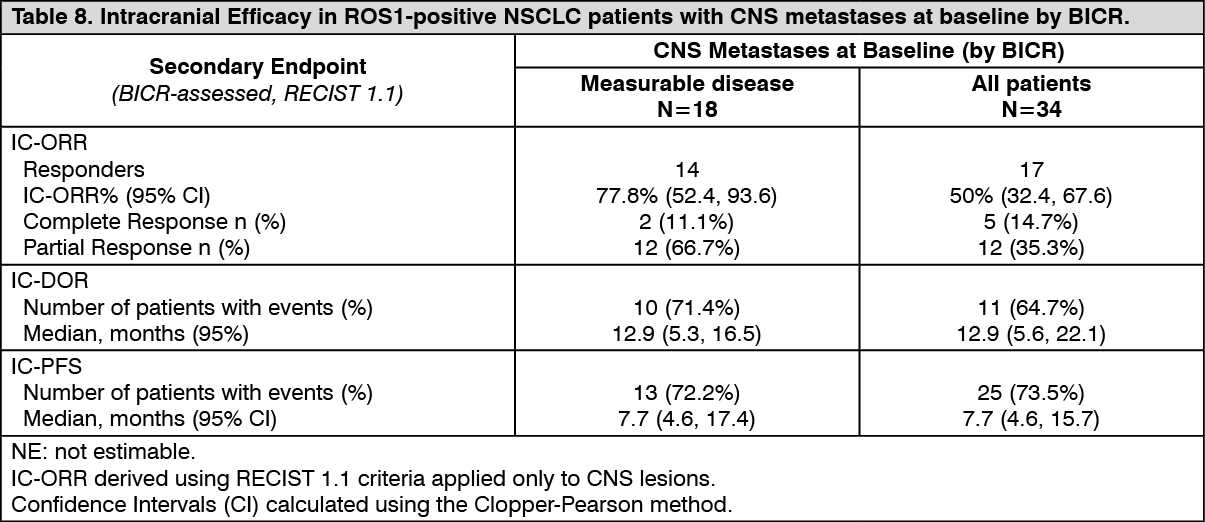 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image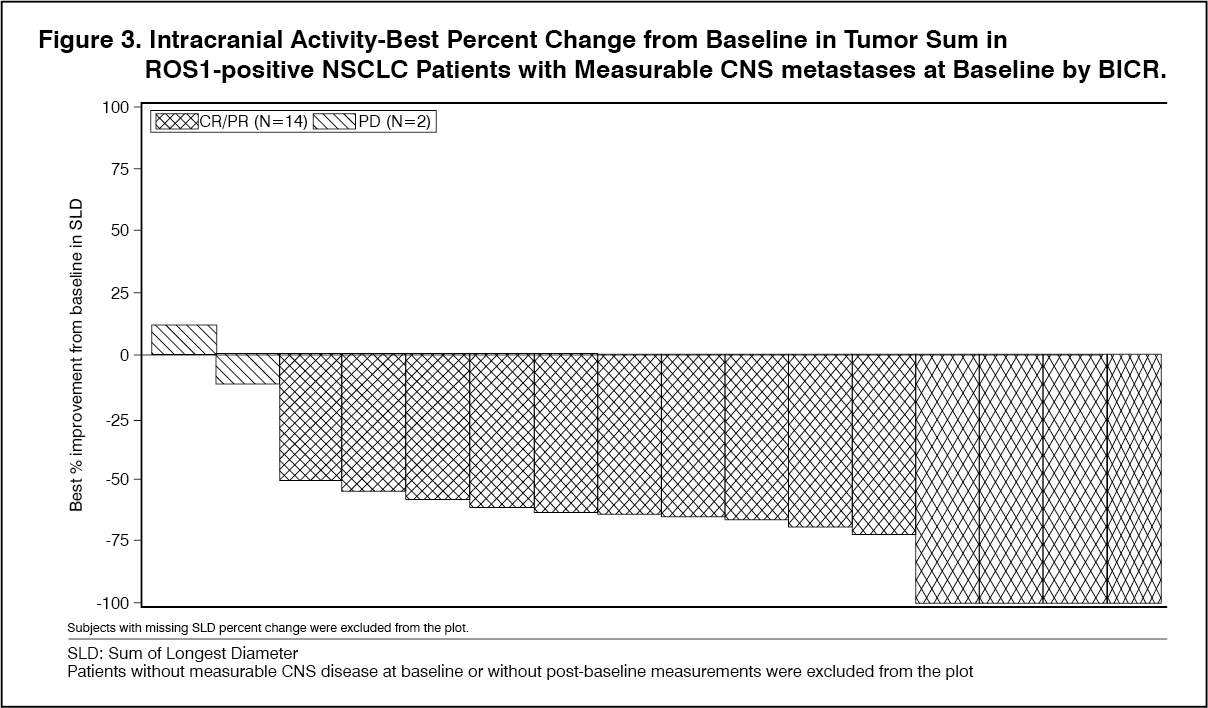 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePatient Reported Outcomes: Patients with ROS1-positive NSCLC reported rapid and durable clinically meaningful improvement (change from baseline of ≥10 points on a 0-100 scale) in lung-cancer symptoms (cough, dyspnea, chest pain) as measured by the EORTC QLQ LC13. Patients maintained their day-to-day function resulting in an improvement in HRQoL while on ROZLYTREK treatment (evaluated by the Physical function, Role function and Global Health Status from the EORTC QLQ-C30). In addition, most patients, indicated that the symptoms commonly associated with ROZLYTREK treatment (lack of appetite, nausea, diarrhea and vomiting) were of low severity, if present.
Immunogenicity: Not applicable.
Pharmacokinetics: The pharmacokinetic parameters for entrectinib and its major active metabolite (M5) have been characterized in patients with NTRK-positive solid tumors and ROS1-positive NSCLC, and healthy subjects.
Following administration of a single 600 mg dose of entrectinib, the estimated entrectinib mean (± SD) Cmax was 1990 (± 1050) nM and AUC0,24 33900 (± 15800) nM*h and for M5 Cmax was 765 (± 598) nM and AUC0,24 13300 (± 10200) nM*h. At steady-state the estimated entrectinib mean Cmax was 3490 (± 1600) nM and AUC0,24 62800 (±29100) nM*h and for M5 Cmax was 1340 (± 934) nM and AUC0,24 25500 (±29100) nM*h.
The population PK model estimated mean accumulation at steady-state following 600 mg once daily administration of entrectinib was 1.89 (±0.381) and 2.01 (± 0.437) for M5.
Absorption: Following a single 600 mg oral administration of ROZLYTREK to patients with NTRK-fusion positive and ROS1 positive NSCLC under fed conditions, entrectinib was rapidly absorbed reaching time-to-maximum plasma concentration (Tmax) after approximately 4 - 6 hours. Based on population pharmacokinetic analysis, steady-state was achieved within 5 days for entrectinib with 600 mg once daily dosing.
The estimated absolute bioavailability of entrectinib based on physiologically based pharmacokinetic (PBPK) modeling was 55%.
No clinically significant effect of food on entrectinib bioavailability was observed. Following a single 600 mg oral administration of ROZLYTREK to healthy subjects under fasting conditions and following a high fat, high calorie meal, the GMR under fed/fasted condition for AUCinf (90%CI) was 115% (107, 124) and for Cmax (90%CI) was 106% (98.9, 115). Entrectinib can be administered with or without food (see Dosage & Administration).
Distribution: Entrectinib and its major active metabolite M5 are highly bound to human plasma proteins independent of drug concentrations. In human plasma, entrectinib and M5 had similar protein binding with >99% bound at a clinically relevant concentration.
After a single oral dose of [14C]-labeled entrectinib, the geometric mean volume of distribution (Vz/F) was 860 L, suggesting extensive distribution into tissues. Population pharmacokinetic analysis estimated volume of distribution of 551 L and 81.1 L for entrectinib and M5, respectively.
Metabolism: Entrectinib is metabolized predominantly by CYP3A4 (~76%). Minor contributions from several other CYPs and UGT1A4 were estimated at <25% in total. The active metabolite M5 (formed by CYP3A4) and the direct N-glucuronide conjugate, M11 (formed by UGT1A4), are the two major circulating metabolites identified.
Elimination: Following administration of a single dose of [14C]-labeled entrectinib administered orally to healthy subjects, the majority of radioactivity was excreted in feces (82.9%) with minimal excretion in urine (3.06%). In feces, 35.7% and 22.1% of the dose was excreted as unchanged entrectinib and M5, respectively, indicating hepatic clearance is the major route of elimination.
Entrectinib and M5 account for approximately 73% of radioactivity in systemic circulation at Cmax, and approximately half of total radioactivity AUCinf.
Population PK analysis estimated a CL/F of 19.6 L/h and 52.4 L/h for entrectinib and M5, respectively. The elimination half-lives of entrectinib and M5 were estimated to be 20 and 40 hours, respectively.
Pharmacokinetics in Special Populations: Pediatric Population: Non-compartmental analysis and population pharmacokinetic modeling approaches demonstrated that the pharmacokinetics of entrectinib and M5 were comparable in adults and children allowing extrapolation of data in adults to pediatric patients.
Data obtained from population pharmacokinetic analyses show that a dose of 300 mg/m2 of ROZLYTREK once daily in pediatric patients results in a similar systemic exposure attained in adults treated with 600 mg of ROZLYTREK, once daily. Population pharmacokinetic analysis data support dosing of pediatric patients with BSA ≥ 1.51 m2 with 600 mg of ROZLYTREK once daily.
Geriatric Population: No differences in entrectinib exposure were noted in patients older than 65 years and younger adults based on pharmacokinetic analysis.
Renal impairment: Negligible amounts of entrectinib and the active metabolite M5 are excreted unchanged in urine (~3% of the dose) indicating renal clearance plays a minor role in the elimination of entrectinib. Population pharmacokinetic data obtained in patients with mild and moderate impairment show that pharmacokinetics of entrectinib are not significantly affected in renal impairment. No formal pharmacokinetic study has been conducted and no population pharmacokinetic data was collected in patients with severe renal impairment. However, since entrectinib elimination via the kidney is negligible, no dose adjustment is required in patients with renal impairment.
Hepatic impairment: The pharmacokinetics of entrectinib were studied in subjects with hepatic impairment due to cirrhosis and subjects with normal hepatic function. Following administration of a single oral dose of 100 mg entrectinib, the AUCinf GMRs (90%CI) of entrectinib were 1.57 (1.03, 2.41) for the mild (Child-Pugh A), 1.54 (1.06, 2.24) for the moderate (Child-Pugh B), and 1.80 (1.22, 2.66) for the severe (Child-Pugh C) hepatic impaired groups compared to the normal hepatic function group. The combined AUClast of entrectinib and M5 showed no relevant change in the hepatic impaired groups compared to the normal hepatic function group. The AUClast GMRs (90%CI) were 1.30 (0.889, 1.89) for the mild, 1.24 (0.886, 1.73) for the moderate, and 1.39 (0.988, 1.95) for the severe hepatic impaired groups compared to the normal hepatic function group.
In addition to the modest increases in entrectinib exposure observed, the variability in systemic exposure was high and observed exposures overlapped across all the study groups.
No dose adjustment is required in patients with underlying mild, moderate or severe hepatic impairment.
Ethnicity: Following a single oral dose of ROZLYTREK in Japanese and Caucasian healthy volunteers, no clinically relevant differences in the exposure of ROZLYTREK were observed. Based on population pharmacokinetics analysis, there was no relationship between systemic exposure of entrectinib and race/ethinicity (Asian, Japanese, white and other ethnicities). No dose adjustment is required for patients of different race/ethnicities. See Special Dosage Instructions under Dosage & Administration.
Toxicology: Preclinical safety data: Carcinogenicity: No carcinogenicity studies have been performed to establish the carcinogenic potential of entrectinib.
Genotoxicity: Entrectinib was not mutagenic in vitro in the bacterial reverse mutation (Ames) assay. Entrectinib was not clastogenic in the in vivo micronucleus assay in rats and did not induce DNA-damage in a comet assay in rats. A potential for abnormal chromosome segregation (aneugenicity) has been detected under in vitro conditions in cultured human peripheral blood lymphocytes (HPBL) but was not detected in the in vivo micronucleus assay in rats.
Impairment of Fertility: No fertility studies in animals have been performed to evaluate the effect of entrectinib. With the exception of dose dependent decreases in prostate weight in male dogs, no effects of entrectinib on reproductive organs were observed in the repeat-dose toxicology studies in rats and dogs at approximately 2.4-fold and 0.6-fold, respectively, the human exposure by AUC at the recommended human dose.
Reproductive toxicity: In an embryo-fetal developmental study in rats, maternal toxicity (decreased body weight gain and food consumption) and fetal malformations (including body closure defects and malformations of the vertebrae and ribs), were observed at 200 mg/kg/day of entrectinib, which represents approximately 2-fold the human exposure by AUC at the recommended dose. Lower fetal weights and reduced skeletal ossification were observed at exposures equivalent to 0.7 times the human exposure by AUC at the recommended dose.
Other: In a 13-week juvenile rat toxicology study from post-natal day 7 to day 97 (approximately equivalent to neonate to 16 years of age in humans), effects on growth and development were observed in the dosing and recovery phases including decreased body weight gain and delayed sexual maturation (at ≥ 4 mg/kg/day, approximately 0.1 times the human exposure by AUC at the recommended dose), deficits in neurobehavioral assessments including functional observational battery and learning and memory (at ≥ 8 mg/kg/day, approximately 0.2 times the human exposure by AUC at the recommended dose) and decreased femur length (at 16 mg/kg/day, approximately 0.3 times the human exposure by AUC at the recommended dose).
Entrectinib penetrates the CNS with brain-to-plasma concentration ratios of ~0.4 in mice, 0.6-1.5 in rats and 1.4-2.2 in dogs following repeated oral daily dosing. Consistent with being a weak P-gp substrate, entrectinib demonstrated high retention in the CNS following IV infusion in rats, achieving sufficient steady-state concentrations in the brain to cover target pharmacological activity at clinically relevant systemic exposure. M5 was also detected in a brain homogenate in rats following a single oral dose or an IV infusion of entrectinib for 5-6 hours, but the exposures of M5 were lower than entrectinib in both plasma and brain in rats.