


 Sign Out
Sign Out

Pharmacology: Mechanism of action: Midostaurin inhibits multiple receptor tyrosine kinases, including FLT3 and KIT kinase. Midostaurin inhibits FLT3 receptor signaling and induces cell cycle arrest and apoptosis in leukemic cells expressing ITD and TKD mutant receptors or overexpressing wild type receptors. Midostaurin inhibits both the wild type and D816V mutant KIT, leading to interference with the aberrant signaling of KIT and inhibits mast cell proliferation and survival, and histamine release.
In addition, it inhibits several other receptor tyrosine kinases such as PDGFR or VEGFR2, as well as members of the serine/threonine kinase family PKC (protein kinase C). Midostaurin binds to the catalytic domain of these kinases and inhibits the mitogenic signaling of the respective growth factors in cells, resulting in growth arrest.
Midostaurin in combination with many chemotherapeutic agents (with the exception of methotrexate) resulted in synergistic growth inhibition in FLT3-ITD expressing AML cell lines.
Pharmacodynamics: Two major metabolites have been identified in murine models and humans.
In proliferation assays with FLT3-ITD expressing cells, CGP62221 showed similar potency compared to the parent compound, whereas CGP52421 was approximately 10 fold less potent.
Cardiac Electrophysiology: A dedicated QT study in 192 healthy subjects with a dose of 75 mg twice daily did not reveal clinically significant prolongation of QT by midostaurin and CGP62221 and the study duration was not long enough to estimate the QTc prolongation effects of the long-acting metabolite CGP52421. Therefore, the change from baseline in QTcF with the concentration of midostaurin and both metabolites was further explored in a phase II study in 116 patients with Advanced SM. At the median peak Cmin concentrations attained at a dose of 100 mg twice daily, neither midostaurin, CGP62221 nor CGP52421 showed a potential to cause clinically significant QTcF prolongation, since the upper bounds of predicted change at these concentration levels were less than 10 msecs with 5.8, 2.4, and 4.0 msecs, respectively.
Clinical Studies: Acute Myeloid Leukemia (AML): The efficacy and safety of Rydapt in combination with standard chemotherapy versus placebo plus standard chemotherapy and as single agent maintenance therapy was investigated in 717 patients (18 to 60 years of age) in a randomized, double-blind, phase III study. Patients with newly diagnosed FLT3 mutated AML as determined by a clinical trial assay were randomized (1:1) to receive Rydapt 50 mg twice daily (n=360) or placebo (n=357) sequentially in combination with standard daunorubicin (60 mg/m2 daily on days 1 to 3)/cytarabine (200 mg/m2 daily on days 1 to 7) induction and high dose cytarabine (3 g/m2 every 12 hours on days 1, 3, 5) consolidation, followed by continuous Rydapt or placebo treatment according to initial assignment for up to 12 additional cycles (28 days/cycle). While the study included patients with various AML related cytogenetic abnormalities, patients with acute promyelocytic leukemia (M3) or therapy related AML were excluded. Patients were stratified by FLT3 mutation status: TKD, ITD with allelic ratio <0.7, and ITD with allelic ratio ≥0.7.
The two treatment groups were generally balanced with respect to the baseline demographics of disease characteristics and details are shown in Table 1. (See Table 1.)
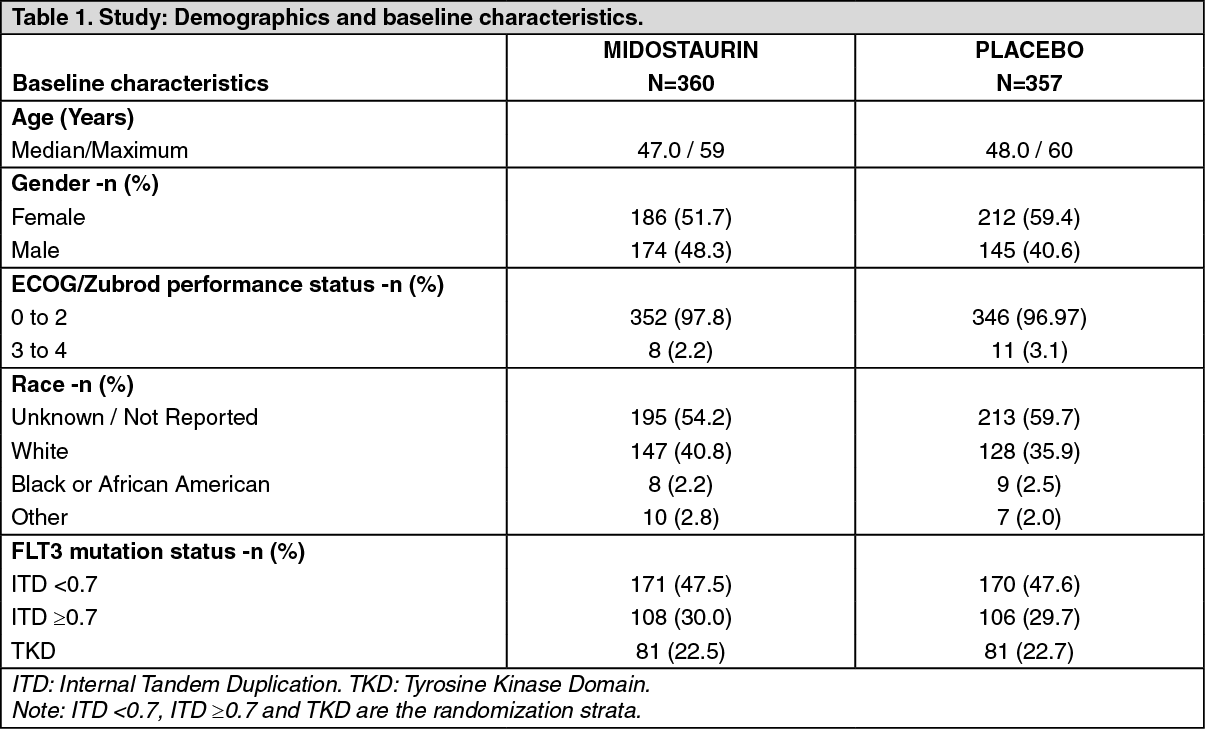 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePatients who proceeded to hematopoietic stem cell transplant (SCT) stopped receiving study treatment on or before the time of stem cell infusion. The overall rate of SCT was 59.4% (214/360) of patient in the Rydapt plus standard chemotherapy arm versus 55.2% (197/357) in the placebo plus standard chemotherapy arm. All patients were followed for survival.
The primary endpoint of the study was overall survival (OS), measured from the date of randomization until death by any cause. The primary analysis was conducted after a minimum follow-up of approximately 3.5 years after the randomization of the last patient. The study demonstrated a statistically significant improvement in OS with a 23% risk reduction of death for Rydapt plus standard chemotherapy over placebo plus standard chemotherapy (Table 2 and Figure 1). (See Figure 1.)
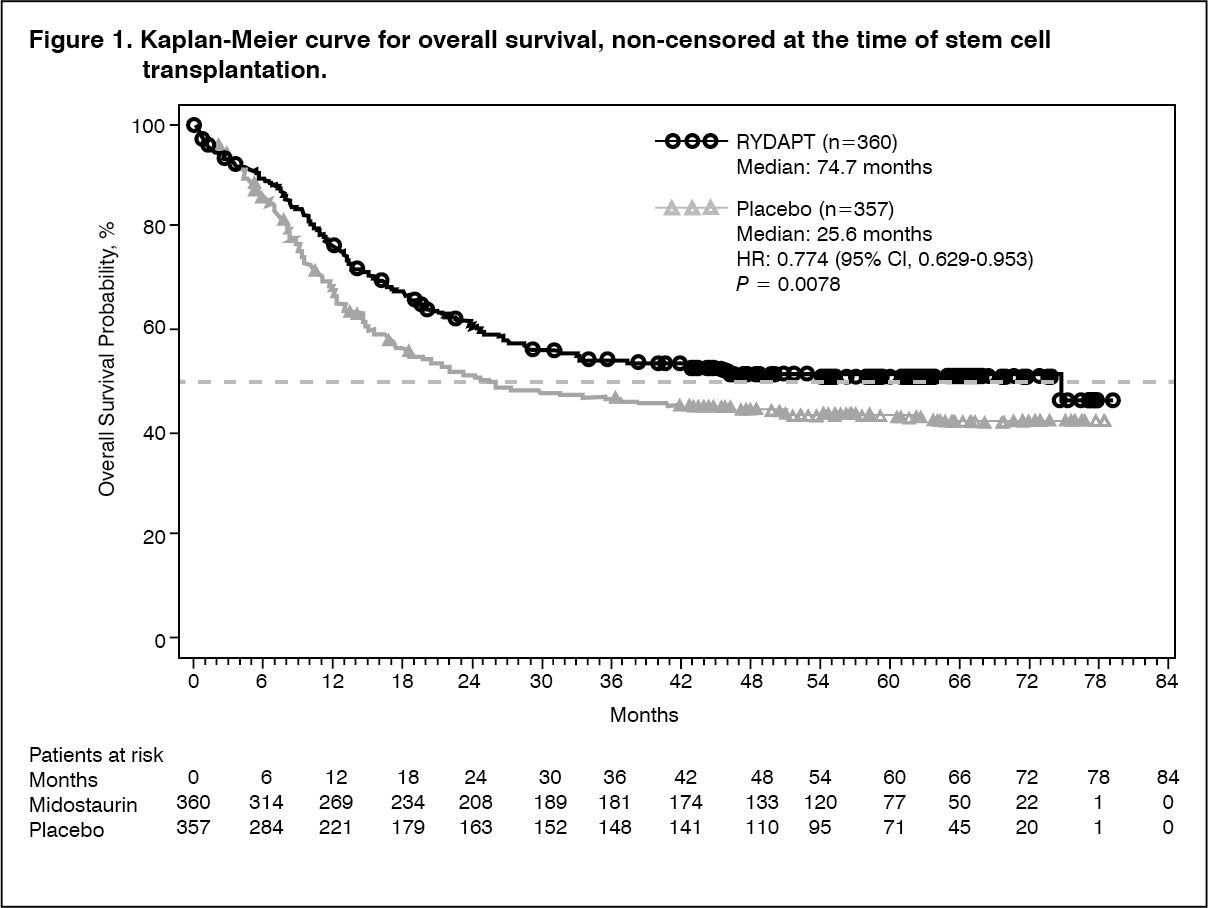 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe key secondary endpoint was event free survival (EFS; an EFS event is defined as a failure to obtain a complete remission (CR) within 60 days of initiation of protocol therapy, or relapse, or death from any cause). The EFS showed a statistically significant improvement for Rydapt plus standard chemotherapy over placebo plus standard chemotherapy (Table 2 and Figure 2). (See Figure 2.)
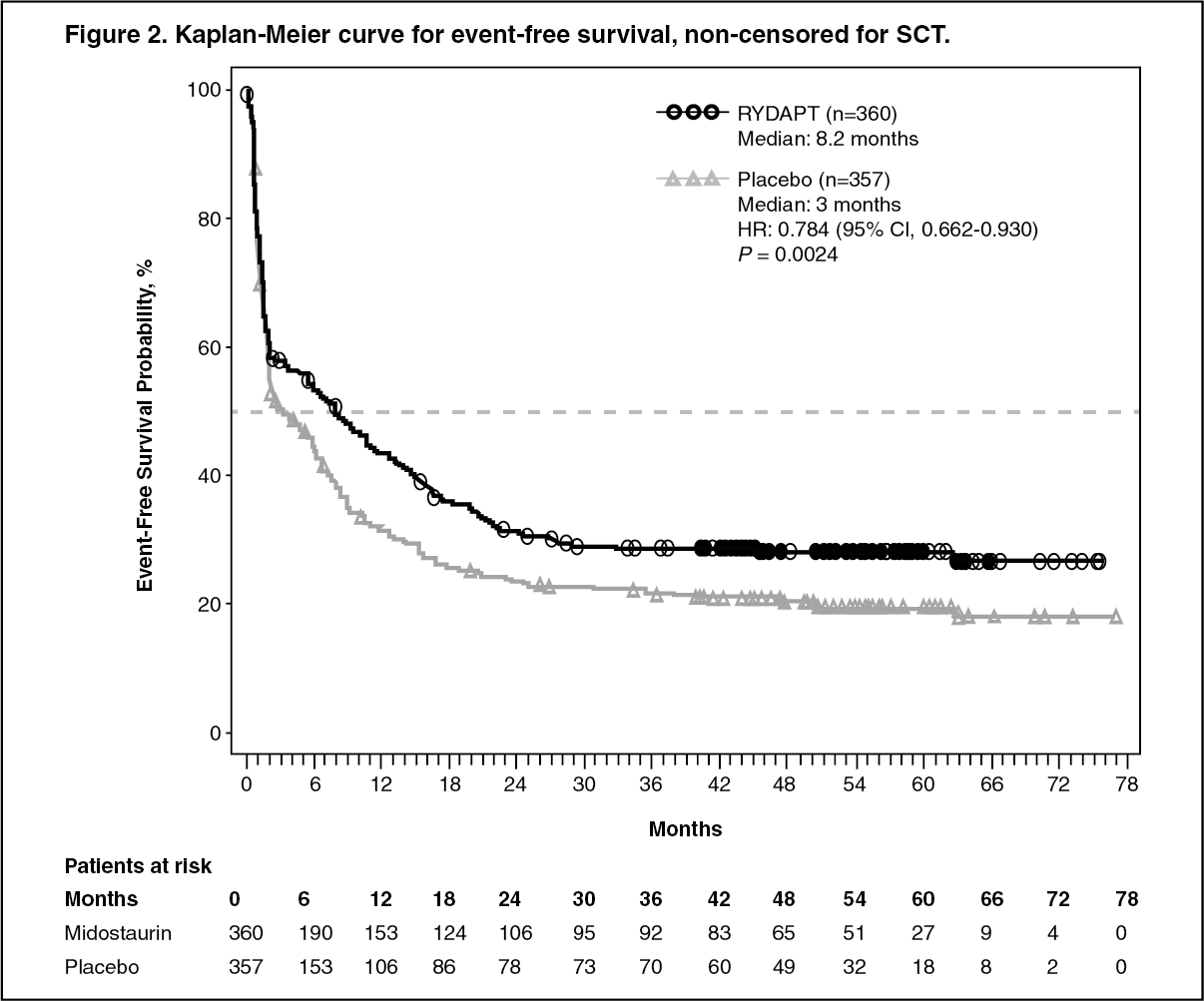 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageSensitivity analyses for both OS and EFS when censored at the time of SCT also supported the clinical benefit with Rydapt plus standard chemotherapy over placebo. There was a trend favoring Rydapt for CR rate by day 60 for the midostaurin arm (58.9% versus 53.5%; P = 0.073) that continued when considering all CRs during induction (65.0% versus 58.0%; P=0.027). In addition, in patients who achieved complete remission in induction, the cumulative incidence of relapse (CIR) at 12 months was 26% in the midostaurin arm vs. 41% in the placebo arm.
Results for OS by SCT status are shown in Figure 3. (See Figure 3 and Table 2.)
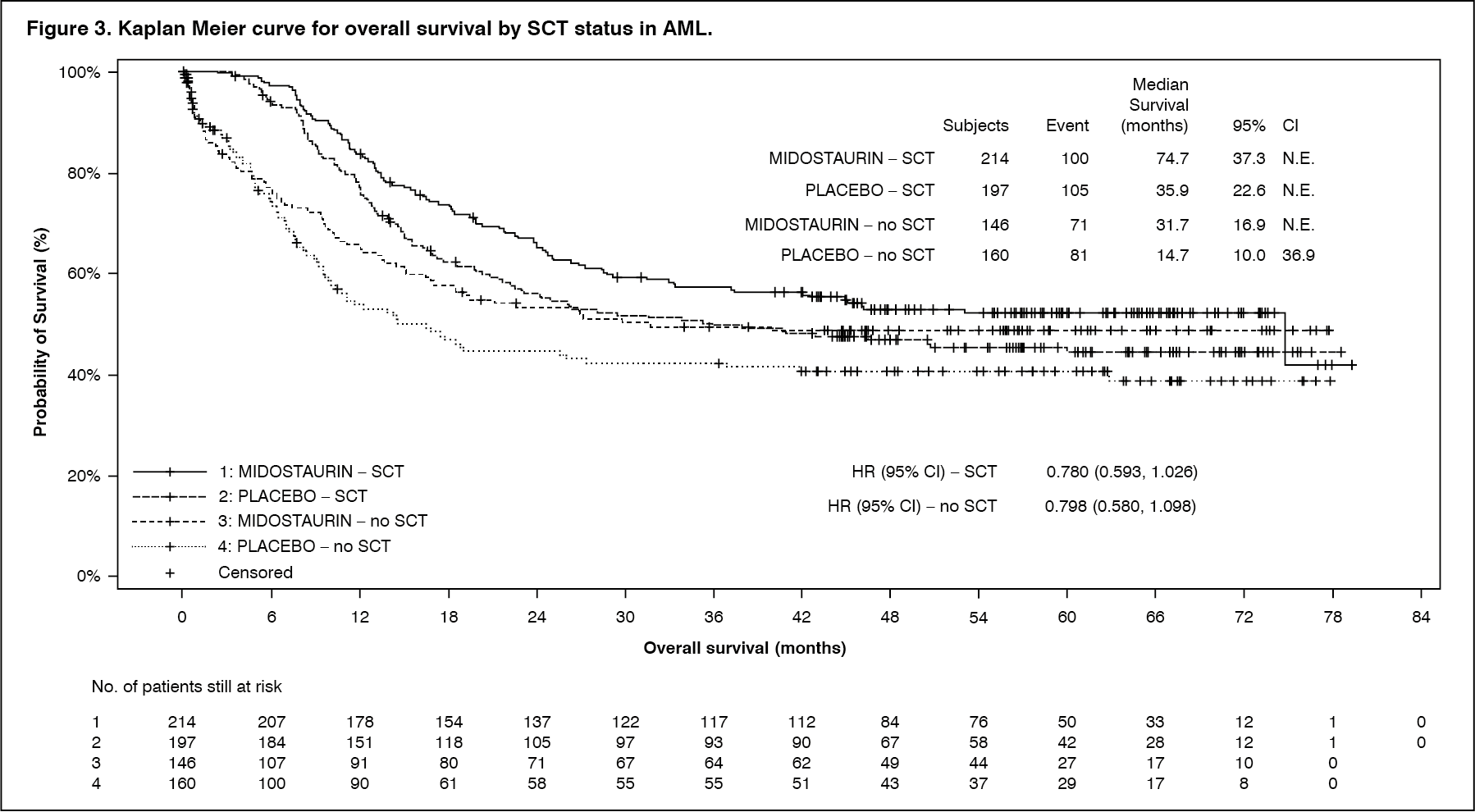 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image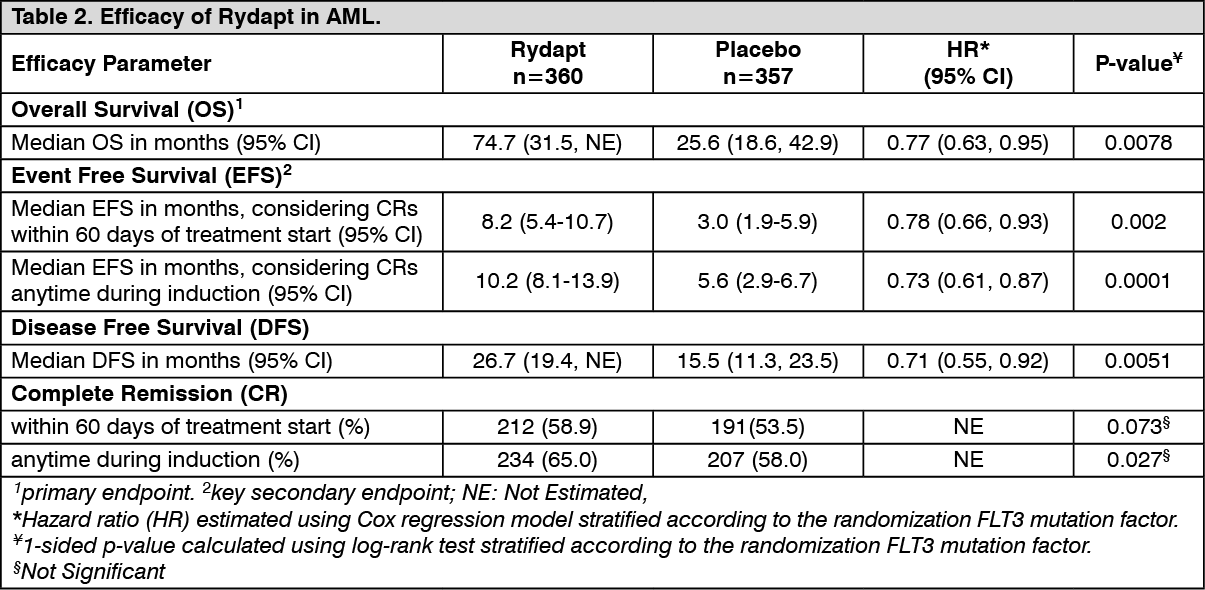 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePediatric patients with AML: In a phase II study, midostaurin was investigated in combination with chemotherapy in newly diagnosed pediatric patients with FLT3-mutated AML. Among the three FLT3-mutated AML patients enrolled in the study, two patients (10 and 14 years old) experienced Dose Limiting Toxicities (DLTs) following the second induction cycle with midostaurin (at 30 mg/m2 twice daily) in combination with chemotherapy (containing cytarabine 2 g/m2/day, day 1 to 5; fludarabine 30 mg/m2/day, day 1 to 5 and idarubicin 12 mg/m2/day, day 2, 4 and 6). Both patients showed markedly delayed hematological recoveries (i.e. prolonged grade 4 thrombocytopenia lasting for 44 days in the first patient and 51 days in the second patient and grade 4 neutropenia lasting for 46 days in the second patient). In the first induction cycle both patients received midostaurin in combination with cytarabine, etoposide and idarubicin.
Advanced Systemic Mastocytosis (ASM): The efficacy of Rydapt in patients with aggressive systemic mastocytosis (ASM) or mast cell leukemia (MCL), with or without an associated hematologic non-mast cell lineage disorder (AHNMD), collectively referred to as Advanced SM, was evaluated in two open-label, single-arm, multicenter studies (142 patients in total).
The pivotal study was a multicenter, single-arm phase II study in 116 patients with Advanced SM (Study CPKC412D2201). Rydapt was administered orally at 100 mg twice daily until disease progression or intolerable toxicity. Of the 116 patients enrolled, 89 were considered eligible for response assessment and constituted the primary efficacy population (PEP). Of these, 73 patients had ASM (57 with an AHNMD), and 16 patients had MCL (6 with an AHNMD). The median age in the PEP was 64 years with approximately half of the patients ≥65 years). Approximately one-third (36%) received prior anti-neoplastic therapy for Advanced SM. At baseline in the PEP, 65% of the patients had >1 measurable C-finding. The KIT D816V mutation was detected in 82% of patients.
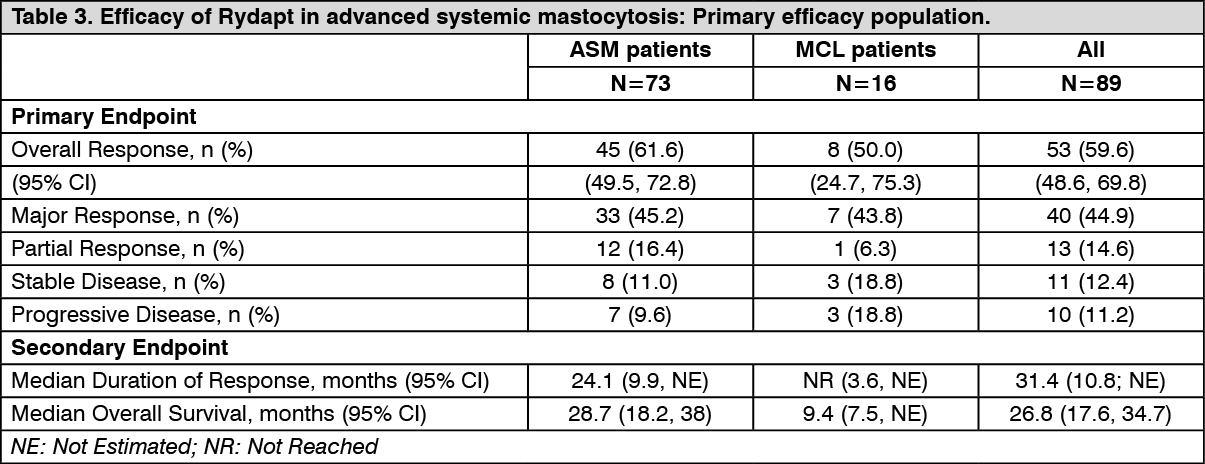
The primary endpoint was overall response rate (ORR). Response rates were assessed based on the modified Valent and Cheson criteria and responses were adjudicated by a study steering committee. Secondary endpoints included duration of response, time to response, and overall survival. Responses to Rydapt are shown in Table 3. Activity was observed regardless of KIT D816V status, number of prior therapies, and presence or absence of an AHNMD. Forty-six percent of patients had a decrease in bone marrow infiltration exceeded 50% and 58% had a decrease in serum tryptase levels exceeded 50%. Spleen volume decreased by ≥10% in 68.9% of patients with at least 1 post-baseline assessment (26.7% of patients had a reduction of ≥35%, which correlates with a 50% decrease by palpation).
The median time to response was 0.3 months (range: 0.1 to 3.7 months). The median duration of follow-up was 43 months. (See Table 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePatient-reported outcome assessments were evaluated using the Memorial Symptom Assessment Scale (MSAS) and SF-12 questionnaires. The most commonly reported baseline symptoms (>65% of prevalence) on the MSAS were "lack of energy", "feeling drowsy", and "difficulty sleeping". The prevalence of all symptoms had decreased at Cycle 12, with the exception of nausea and vomiting. Maximum improvement was reported for the symptom "weight loss", where the prevalence decreased from 50% to 17%. A similar pattern of improvements was seen at Cycle 6, and for best TMSAS score during the study. Overall, responders showed more improvement than non-responders with no worsening of any symptoms. For the SF-12 physical component score, best mean score at baseline were below those reported for a healthy population, whereas best mean scores reported during the study approached those reported for a healthy population, especially in responders. A similar trend was observed for the SF-12 mental component scale.
The supportive study was a single arm, multicenter, open-label phase II study of 26 patients with Advanced SM (CPKC412A2213). Rydapt was administered orally at 100 mg twice daily. Lack of a major response (MR) or partial response (PR) by the end of the second cycle resulted in discontinuation from the study treatment. Twenty (76.9%) patients had ASM (17 [85%] with AHNMD) and 6 patients (23.1%) had MCL (2 [33.3%] with AHNMD). The median age was 64.5 years with half of the patients ≥65 years. At baseline, 88.5% had >1 C-finding and 69.2% had received at least one prior anti-neoplastic regimen.
The primary endpoint was ORR evaluated by the Valent criteria during the first two cycles of treatment. Nineteen patients (73.1%; 95% CI=[52.2, 88.4]) achieved a response during the first two cycles of treatment (13 MR; 6 PR). The median duration of follow-up was 73 months, and the median duration of response has not been reached. Median overall survival was 40.0 months (patients were only followed for up one year after treatment discontinuation for survival).
Pharmacokinetics: Absorption: In humans, the absorption of midostaurin is rapid after oral administration, with Tmax of total radioactivity observed at 1 to 3 hours post dose. In healthy subjects, the extent of midostaurin absorption (AUC) was increased by an average of 22% when Rydapt was co-administered with a standard meal, and by an average of 59% when co-administered with a high-fat meal. Peak midostaurin concentration (Cmax) was reduced by 20% with a standard meal and by 27% with a high-fat meal versus on an empty stomach. Time to peak concentration were also delayed in presence of a standard meal or a high-fat meal (median Tmax=2.5 hrs to 3 hrs). In clinical studies, midostaurin was administered with a light meal, in order to decrease potential nausea and vomiting events and it is recommended that midostaurin is administered to patients with food.
Distribution: Midostaurin has a high tissue distribution of geometric mean Vz/F=95.2 L. Midostaurin and its metabolites are distributed mainly in plasma rather than red blood cells. In vitro data showed midostaurin is greater than 98% bound to plasma protein mainly to alpha-1-acid glycoprotein (AGP).
Biotransformation/metabolism: Midostaurin is metabolized by CYP3A4 mainly via oxidative pathways and the major plasma components included midostaurin and two major active metabolites; CGP62221 and CGP52421 accounting for 27.7±2.7% and 37.97±6.6% respectively of the total plasma exposure.
Elimination: The median terminal half-lives of midostaurin, CGP62221 and CGP52421 in plasma are approximately 20.9, 32.3 and 471 hours. The Human Mass Balance study results indicate that fecal excretion is the major route of excretion (78% of the dose), and mostly as metabolites (73% of the dose) while unchanged midostaurin accounts for 3% of the dose. Only 4% of the dose is recovered in urine.
Linearity/non-linearity: In general, midostaurin and its metabolites showed no major deviation from dose-proportionality after a single dose in the range of 25 mg to 100 mg. However, there was a less than dose-proportional increase in exposure after multiple doses within the dose range of 50 mg to 225 mg daily.
Following multiple oral doses, midostaurin displayed time-dependent pharmacokinetics with an initial increase in plasma concentrations during the first week (peak Cmin) followed by a decline with time to a steady-state after approximately 28 days. While the exact mechanism for the declining concentration of midostaurin is unclear, it may be possibly due to CYP3A4 enzyme auto-induction. The pharmacokinetics of the CGP62221 metabolite showed a similar trend. However, CGP52421 concentrations increased up to 2.5 fold with Advanced SM to and up to 9-fold for AML, compared to midostaurin after one month of treatment.
In vitro evaluation of drug interaction potential: Based on in vitro data, midostaurin and its active metabolites, CGP52421 and CGP62221, are considered inhibitors of CYP1A2 and CYP2E1 and inducers of CYP2B6 (induction mediated by CAR) and CYP1A2 (induction mediated by AhR).
In vitro experiments demonstrated that midostaurin, CGP52421 and CPG62221 can potentially inhibit BCRP and BSEP. Simulations using physiologically-based pharmacokinetic (PBPK) models predicted that midostaurin given at a dose of 50 mg or 100 mg twice daily at steady state is unlikely to cause clinically relevant inhibition of OATP1B.
Special populations: Pediatric patients (below 18 years): The pharmacokinetics of midostaurin in pediatric patients were explored in a phase I dose escalation monotherapy study with 22 patients (ages 3 months to 18 years of age) with AML or MLL-rearranged ALL using a population PK approach. After adjusting for body weight, exposures of midostaurin and its two metabolites in pediatrics fell within the ranges predicted by modeling data from adults.
Geriatric patients (65 years or above): Based on population PK model analyses of the effect of age on clearance of midostaurin and its active metabolites, there was no statistically significant finding and the anticipated changes in exposure were not deemed to be clinically relevant. In adult patients with Advanced SM or AML, no midostaurin dose adjustment is required based on age.
Gender: Based on population PK model analyses of the effect of gender on clearance of midostaurin and its active metabolites, there was no statistically significant finding and the anticipated changes in exposure were not deemed to be clinically relevant. No midostaurin dose adjustment is required based on gender.
Race/Ethnicity: There are no differences in the pharmacokinetic profile between Caucasian and Black subjects. Based on the phase I study in healthy Japanese volunteers, pharmacokinetic profiles of midostaurin and its metabolites (CGP62221 and CGP52421) are similar compared to those observed in other PK studies conducted in Caucasians and Blacks. No midostaurin dose adjustment is required based on ethnicity.
Patients with hepatic impairment: A dedicated hepatic impairment study assessed the systemic exposure of midostaurin after oral administration of 50 mg twice daily for 6 days and a single 50 mg dose on day 7 in subjects with baseline mild or moderate (Child-Pugh Class A or B, respectively) and following a single dose administration of 50 mg in subjects with severe hepatic impairment (Child-Pugh Class C) in comparison to control subjects with normal hepatic function. The maximum concentration of midostaurin was reached between 2 and 3 hours after administration after single or repeated doses for all groups. On day 1, the AUC0-12 and Cmax were 8130 ng*h/ml and 1206 ng/ml, respectively, for healthy subjects. AUC0-12 was decreased by 39% and 36% in subjects with mild and moderate hepatic impairment, respectively. On day 7, AUCCtrough (exposure under the curve of Ctrough from day 1 to day 7) was 5410 ng*h/ml in healthy subjects and was decreased by 35% and 20% in subjects with mild and moderate hepatic impairment, respectively. AUCtau was decreased by 28% and 20% on day 7, respectively.
The subjects with severe hepatic impairment had a lower geometric mean Cmax and AUCinf of midostaurin compared to the control group (Cmax: 1360 ng/ml, AUCinf: 30100 ng·h/ml). Cmax and AUCinf of midostaurin decreased on average by 78% and 59% respectively in subjects with severe hepatic impairment.
Finally, the long-term data from patients were analysed using a population pharmacokinetic approach. No impact of hepatic impairment could be identified in patients with mild or moderate hepatic impairment in the ASM, SM-AHN, MCL and AML populations.
Overall, there was no increase in exposure (AUC) to plasma midostaurin and its metabolites (CGP62221 and CGP52421) in subjects with mild, moderate or severe hepatic impairment compared to subjects with normal hepatic function. No dosage adjustment is necessary for patients with baseline mild or moderate hepatic impairment. Exposure to midostaurin and its active metabolite CGP62221 is substantially lower in patients with severe hepatic impairment than that in patients with normal hepatic function (see Dosage & Administration). However, there are insufficient efficacy data in patients with severe hepatic impairment to suggest a dose adjustment is required.
Patients with renal impairment: No dedicated renal impairment study was conducted for midostaurin. Population pharmacokinetic (popPK) analyses were conducted using data from clinical trials in patients with AML (n=180) and Advanced SM (n=141). Out of the 321 patients included, 177 patients showed pre-existing mild (n=113), moderate (n=60) or severe (n=4) renal impairment (15 mL/min ≤creatinine clearance [CrCL] <90 mL/min). 144 patients showed normal renal function (CrCL >90 mL/min) at baseline. Based on the population PK analyses, midostaurin clearance was not significantly impacted by renal impairment and therefore, no dosage adjustment is necessary for patients with mild or moderate renal impairment.
Toxicology: Non-clinical safety data: Midostaurin has been evaluated in safety pharmacology, single/repeated dose toxicity, genotoxicity, reproductive and developmental toxicity studies.
Safety pharmacology and single/repeat dose toxicity: Safety pharmacology studies indicate that midostaurin is unlikely to interfere with vital functions of the central nervous systems. In vitro, midostaurin did not inhibit hERG channel activity up to the limit of solubility of 12 microM. The two major human metabolites CGP52421 and CGP62221 (also tested up to the limit of solubility) inhibited hERG current by 38.5% at 1.5 microM and 11.3% at 1.21 microM respectively. Midostaurin and the two metabolites are highly protein bound and the free concentrations at therapeutic doses are far below the concentrations associated with no/minimal hERG inhibition in vitro. The risk of hERG related-QT prolongation appears to be low. In the repeat dose studies in dogs, a decrease in heart rate and a prolongation of the P-Q interval was seen in individual animals at 10 and 30 mg/kg; there were no morphological changes in the heart.
In the repeat dose studies, the key target organs identified were the gastrointestinal tract (emesis in dogs and monkeys, diarrhea and mucosal alteration), testes (decreased spermatogenesis), bone marrow (hypocellularity) and lymphoid organs (depletion/atrophy). The effect on the bone marrow and lymphoid organs was accompanied by hematological changes of decreased white blood cells, lymphocytes and erythrocytic parameters. An increase in liver enzymes (ALT and AST) was seen consistently in rats, and in dogs and monkeys in long term studies of ≥3 months duration. There were no corresponding pathological changes in the liver. Inhibition of spermatogenesis was seen in dogs at doses ≥3 mg/kg. The no-adverse-effect level after 12 months of treatment was 1 mg/kg in dogs and 3 mg/kg in rats.
Reproductive toxicity: See Use in Pregnancy & Lactation.
Juvenile animal studies: In a toxicity study in juvenile rats, midostaurin was administered at 2, 5 and 15 mg/kg/day from days 7 to 70 postpartum. A reduction in body weight, hemorrhage, mixed cell infiltration in the lungs and erythrocytosis/erythrophagocytosis in the mesenteric lymph nodes were seen at 15 mg/kg/day. There were no effects on physical development, sensory function, behavioral or reproductive function. The no-observed-adverse-effect level was 5 mg/kg/day.
Genotoxicity: In vitro and in vivo genotoxicity studies covering relevant genotoxicity endpoints showed no evidence of mutagenic or clastogenic activity. No carcinogenicity studies have been performed.