


 Sign Out
Sign Out
In placebo-controlled and uncontrolled clinical studies, a total of 2,468 patients have received Tecfidera and been followed for periods up to 4 years with an overall exposure equivalent to 3,588 person-years. Approximately 1,056 patients have received more than 2 years of treatment with Tecfidera. The experience in uncontrolled clinical trials is consistent with the experience in the placebo-controlled clinical trials.
Tabulated summary of adverse reactions: Adverse reactions, which were more frequently reported in Tecfidera versus placebo-treated patients, are presented in the table below. These data were derived from 2 pivotal Phase 3 placebo-controlled, double-blind clinical trials with a total of 1,529 patients treated with Tecfidera and for up to 24 months with an overall exposure of 2,371 person-years (see Pharmacology: Pharmacodynamics under Actions). The frequencies described in the table below are based on 769 patients treated with Tecfidera 240 mg twice a day and 771 patients treated with placebo.
The adverse reactions are presented as MedDRA preferred terms under the MedDRA System Organ Class. The incidence of the adverse reactions below are expressed according to the following categories: Very common (≥1/10), Common (≥1/100 to <1/10), Uncommon (≥1/1, 000 to <1/100), Rare (≥1/10, 000 to <1/1,000), Very rare (<1/10,000), Not known (frequency cannot be estimated from the available data). (See Table 2.)
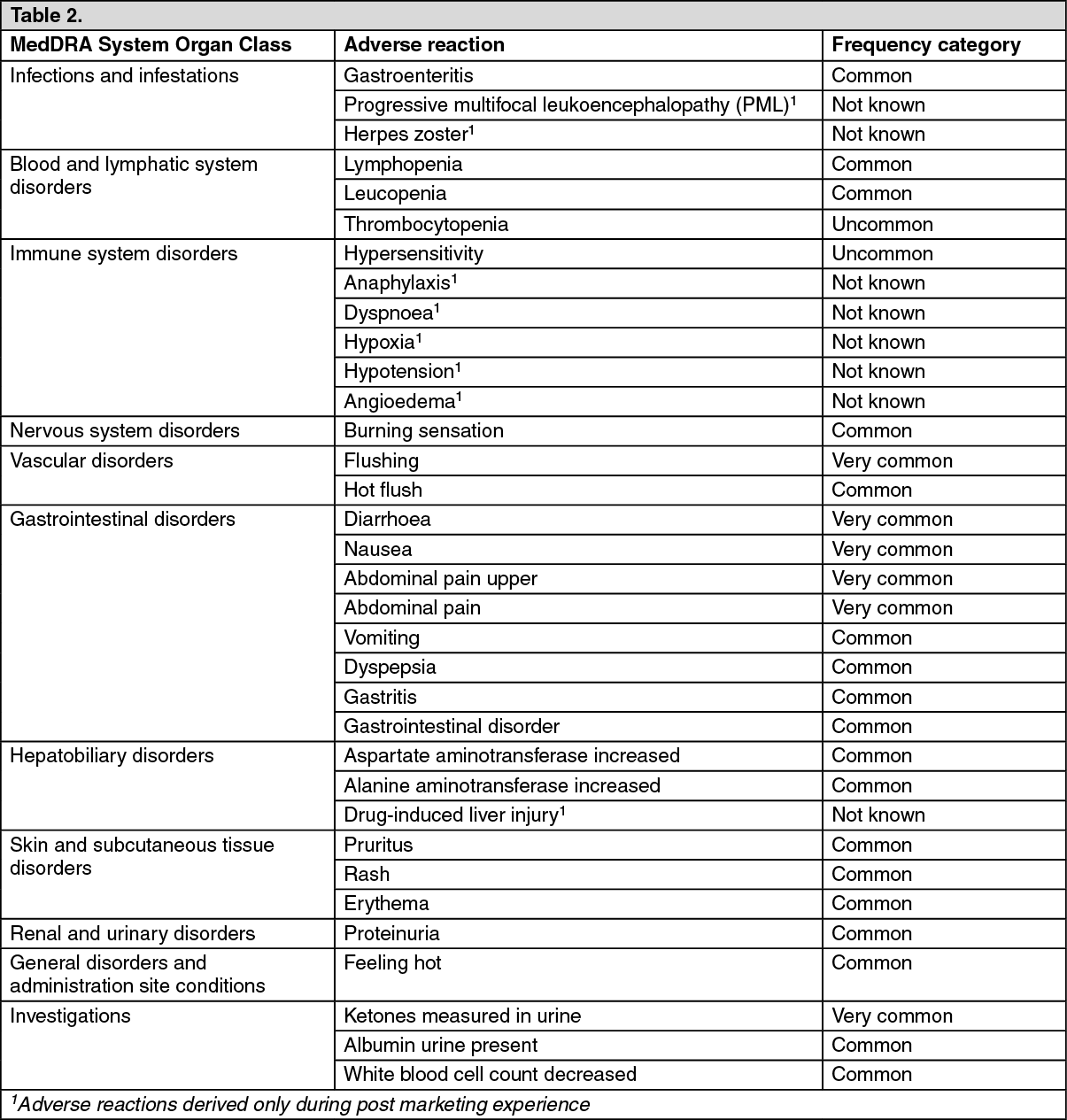 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageDescription of selected adverse reactions: Flushing: In the placebo-controlled studies, the incidence of flushing (34% versus 4%) and hot flush (7% versus 2%) was increased in patients treated with Tecfidera compared to placebo, respectively. Flushing is usually described as flushing or hot flush, but can include other events (e.g. warmth, redness, itching, and burning sensation). Flushing events tend to begin early in the course of treatment (primarily during the first month) and in patients who experience flushing, these events may continue to occur intermittently throughout treatment with Tecfidera. In patients with flushing, the majority had flushing events that were mild or moderate in severity. Overall, 3% of patients treated with Tecfidera discontinued due to flushing. The incidence of serious flushing, which may be characterised by generalised erythema, rash and/or pruritus, was seen in less than 1% of patients treated with Tecfidera (see Dosage & Administration, Precautions and Interactions).
Gastrointestinal: The incidence of gastrointestinal events (e.g. diarrhoea [14% versus 10%], nausea [12% versus 9%], upper abdominal pain [10% versus 6%], abdominal pain [9% versus 4%], vomiting [8% versus 5%] and dyspepsia [5% versus 3%]) was increased in patients treated with Tecfidera compared to placebo, respectively. Gastrointestinal events tend to begin early in the course of treatment (primarily during the first month) and in patients who experience gastrointestinal events, these events may continue to occur intermittently throughout treatment with Tecfidera. In the majority of patients who experienced gastrointestinal events, it was mild or moderate in severity. Four per cent (4%) of patients treated with Tecfidera discontinued due to gastrointestinal events. The incidence of serious gastrointestinal events, including gastroenteritis and gastritis, was seen in 1% of patients treated with Tecfidera (see Dosage & Administration).
Hepatic function: Based on data from placebo-controlled studies, the majority of patients with elevations had hepatic transaminases that were <3 times the upper limit of normal (ULN). The increased incidence of elevations of hepatic transaminases in patients treated with Tecfidera relative to placebo was primarily seen during the first 6 months of treatment. Elevations of alanine aminotransferase and aspartate aminotransferase ≥3 times ULN, respectively, were seen in 5% and 2% of patients treated with placebo and 6% and 2% of patients treated with Tecfidera. Discontinuations due to elevated hepatic transaminases were <1% and similar in patients treated with Tecfidera or placebo. Elevations in transaminases ≥3 times ULN with concomitant elevations in total bilirubin >2 times ULN, were not observed in placebo-controlled studies.
Increase of liver enzymes and cases of drug-induced liver injury (elevations in transaminases ≥3 times ULN with concomitant elevations in total bilirubin >2 times ULN), have been reported in post marketing experience following Tecfidera administration, which resolved upon treatment discontinuation.
Infections: Herpes zoster infections have been reported with Tecfidera use. In an ongoing long-term extension study, in which 1736 MS patients are treated with Tecfidera, approximately 5% experienced one or more events of herpes zoster, the majority of which were mild to moderate in severity. Most subjects, including those who experienced a serious herpes zoster infection, had lymphocyte counts above the lower limit of normal. Grade 2 and 3 lymphopenia prevailed in subjects with concurrent lymphocytopenia. In the post-marketing setting most cases of herpes zoster infection were non-serious and resolved with treatment. Limited data is available on ALC in patients with herpes zoster infection in the post-marketing setting. However, when reported, most patients experienced grade 2 (< 0.8 × 109/L to 0.5 × 109/L) or grade 3 (<0.5 × 109/L to 0.2 × 109/L) lymphopenia (see Precautions).
Lymphopenia: In the placebo-controlled studies most patients (>98%) had normal lymphocyte values prior to initiating treatment. Upon treatment with Tecfidera, mean lymphocyte counts decreased over the first year with a subsequent plateau. On average, lymphocyte counts decreased by approximately 30% of baseline value. Mean and median lymphocyte counts remained within normal limits. Lymphocyte counts <0.5x109/l were observed in <1% of patients treated with placebo and 6% of patients treated with Tecfidera. A lymphocyte count <0.2x109/l was observed in 1 patient treated with Tecfidera and in no patients treated with placebo. In clinical studies (both controlled and uncontrolled), 9% of patients had lymphocyte counts ≥0.5 x 109/L and <0.8 x 109/L for at least six months; 2% of patients experienced lymphocyte counts <0.5 x 109/L for at least six months, and in this group the majority of lymphocyte counts remained <0.5 x 109/L with continued therapy.
PML has occurred in the setting of moderate to severe prolonged lymphopenia (see Precautions).
Laboratory abnormalities: In the placebo-controlled studies, measurement of urinary ketones (1+ or greater) was higher in patients treated with Tecfidera (45%) compared to placebo (10%). No untoward clinical consequences were observed in clinical trials.
Levels of 1,25-dihydroxyvitamin D decreased in Tecfidera treated patients relative to placebo (median percentage decrease from baseline at 2 years of 25% versus 15%, respectively) and levels of parathyroid hormone (PTH) increased in Tecfidera treated patients relative to placebo (median percentage increase from baseline at 2 years of 29% versus 15%, respectively). Mean values for both parameters remained within normal range.
A transient increase in mean eosinophil counts was seen during the first 2 months of therapy.
Paediatric population: The safety of Tecfidera in paediatric patients with multiple sclerosis below the age of 18 has not yet been established. In a small 24-week open-label uncontrolled study in paediatric patients with RRMS aged 13 to 17 years (120 mg twice a day for 7 days followed by 240 mg twice a day for the remainder of treatment; safety population, n=22), followed by a 96 week extension study (240mg twice per day; safety population n=20), the safety profile appeared similar to that observed in adult patients.
Reporting of suspected adverse reactions: Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system.
View ADR Monitoring Form