Therapeutic/Pharmacologic Class of Drug: Antineoplastic agents, monoclonal antibodies.
ATC Code: L01FA01.
PHARMACOLOGY: Pharmacodynamics: Mechanism of Action: Rituximab is a chimeric mouse/human monoclonal antibody that binds specifically to the transmembrane antigen CD20. This antigen is located on pre-B- and mature B-lymphocytes, but not on haemopoietic stem cells, pro-B-cells, normal plasma cells or other normal tissue. The antigen is expressed on >95% of all B-cell non-Hodgkin's lymphomas (NHLs). Following antibody binding, CD20 is not internalized or shed from the cell membrane into the environment. CD20 does not circulate in the plasma as a free antigen and, thus, does not compete for antibody binding.
Rituximab binds to the CD20 antigen on B-lymphocytes and initiates immunologic reactions that mediate B-cell lysis. Possible mechanisms of cell lysis include complement-dependent cytotoxicity (CDC), antibody-dependent cellular cytotoxicity (ADCC), and induction of apoptosis. Finally, in vitro studies have demonstrated that rituximab sensitizes drug-resistant human B-cell lymphoma lines to the cytotoxic effects of some chemotherapeutic agents.
Peripheral B-cell counts declined to levels below normal following the first dose of rituximab. In patients treated for haematological malignancies, B-cell recovery began within 6 months of treatment returning to normal levels within 6 months of treatment and generally returning to normal levels, within 12 months after completion of therapy, although in some patients this may take longer (see Clinical Trials: Experience from Clinical Trials in Haemato-Oncology under Adverse Reactions).
In patients with rheumatoid arthritis, the duration of peripheral B cell depletion was variable. The majority of patients received further treatment prior to full B cell repletion. A small proportion of patients had prolonged peripheral B-cell depletion lasting 2 years or more after their last dose of rituximab.
In GPA and MPA patients, peripheral blood CD19 B-cells depleted to less than 10 cells/µl following the first two infusions of rituximab and remained at that level in most patients through month 6.
Of 67 patients evaluated for human anti-mouse antibody (HAMA), none were positive. Of 356 non-Hodgkin's lymphoma patients evaluated for human anti-chimeric antibody (HACA) 1.1% (4 patients) were positive.
Clinical/Efficacy Studies: Low-grade or follicular non-Hodgkin's lymphoma: Monotherapy: Initial treatment, weekly for 4 doses: In the pivotal study, 166 patients with relapsed or chemoresistant low-grade or follicular B-cell NHL received 375 mg/m
2 of rituximab as an IV infusion weekly for four doses. The overall response rate (ORR) in the intent-to-treat (ITT) population was 48% (CI
95% 41% - 56%) with a 6% complete response (CR) and a 42% partial response (PR) rate. The projected median time to progression (TTP) for responding patients was 13.0 months.
In a subgroup analysis, the ORR was higher in patients with IWF B, C, and D histologic subtypes as compared to IWF A subtype (58% versus 12%), higher in patients whose largest lesion was < 5 cm versus > 7 cm in greatest diameter (53% versus 38%), and higher in patients with chemosensitive relapse as compared to chemoresistant (defined as duration of response < 3 months) relapse (50% versus 22%). ORR in patients previously treated with autologous bone marrow transplant (ABMT) was 78% versus 43% in patients with no ABMT. Neither age, sex, lymphoma grade, initial diagnosis, presence nor absence of bulky disease, normal or high LDH nor presence of extranodal disease had a statistically significant effect (Fisher's exact test) on response to rituximab. A statistically significant correlation was noted between response rates and bone marrow involvement. Forty percent of patients with bone marrow involvement responded compared to 59% of patients with no bone marrow
involvement (p = 0.0186). This finding was not supported by a stepwise logistic regression analysis in which the following factors were identified as prognostic factors: histologic type, bcl-2 positivity at baseline, resistance to last chemotherapy and bulky disease.
Initial treatment, weekly for 8 doses: In a multi-center, single-arm study, 37 patients with relapsed or chemoresistant, low grade or follicular B-cell NHL received 375 mg/m
2 of rituximab as IV infusion weekly for eight doses. The ORR was 57% (CI
95% 41% - 73%; CR 14%, PR 43%) with a projected median TTP for responding patients of 19.4 months (range 5.3 to 38.9 months).
Initial treatment, bulky disease, weekly for 4 doses: In pooled data from three studies, 39 patients with relapsed or chemoresistant, bulky disease (single lesion ≥ 10 cm in diameter), low grade or follicular B-cell NHL received 375 mg/m
2 of rituximab as IV infusion weekly for four doses. The ORR was 36% (CI
95% 21% - 51%; CR 3%, PR 33%) with a median TTP for responding patients of 9.6 months (range 4.5 to 26.8 months).
Re-treatment, weekly for 4 doses: In a multicenter, single-arm study, 58 patients with relapsed or chemoresistant low grade or follicular B-cell NHL, who had achieved an objective clinical response to a prior course of rituximab, were re-treated with 375 mg/m
2 of rituximab as IV infusion weekly for four doses. Three of the patients had received two courses of rituximab before enrollment and thus were given a third course in the study. Two patients were re-treated twice in the study. For the 60 re-treatments on study, the ORR was 38% (CI
95% 26% - 51%; 10% CR, 28% PR) with a projected median TTP for responding patients of 17.8 months (range 5.4 - 26.6). This compares favorably with the TTP achieved after the prior course of rituximab (12.4 months).
Rituximab in combination with chemotherapy: Initial treatment: In an open-label randomized trial, a total of 322 previously untreated patients with follicular lymphoma were randomized to receive either CVP chemotherapy (cyclophosphamide 750 mg/m
2, vincristine 1.4 mg/m
2 up to a maximum of 2 mg on day 1, and prednisolone 40 mg/m
2/day on days 1 - 5) every 3 weeks for 8 cycles or rituximab 375 mg/m
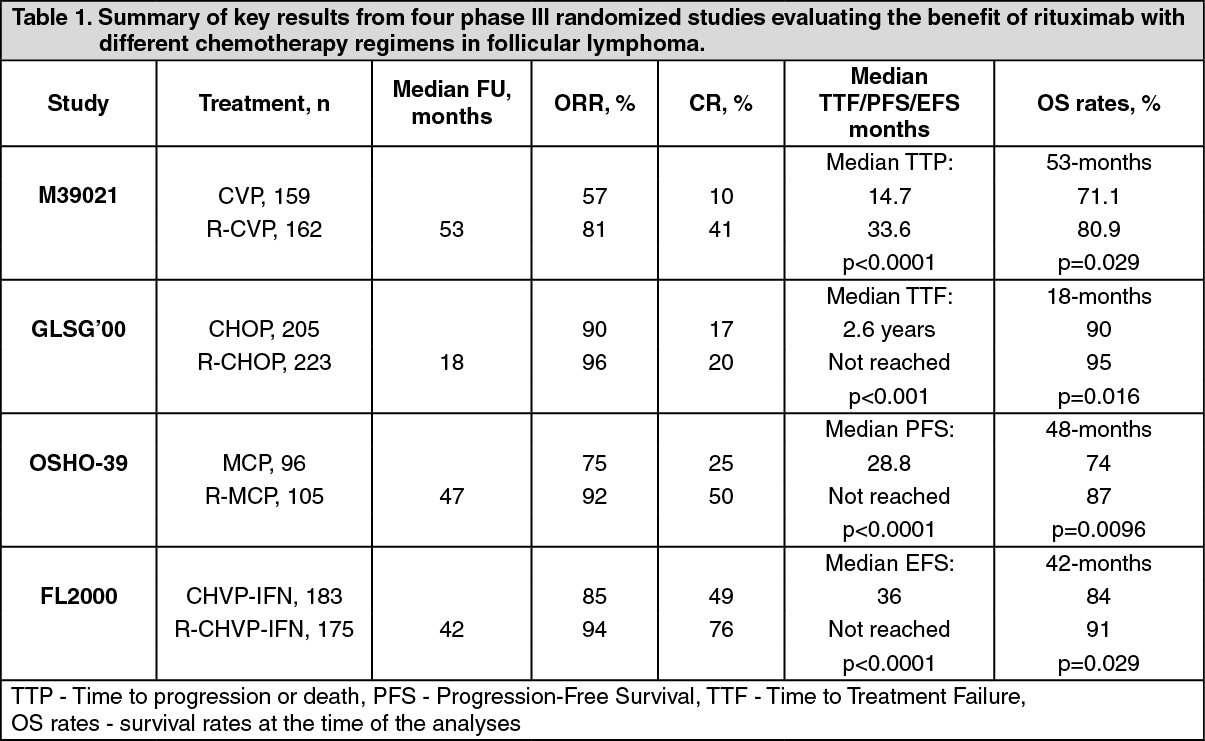
2 in combination with CVP (R-CVP). Rituximab was administered on the first day of each treatment cycle. A total of 321 patients (162 R-CVP, 159 CVP) received therapy and were analyzed for efficacy. The median follow-up of patients was 53 months, R-CVP led to a significant benefit over CVP for the primary endpoint, time to treatment failure (27 months versus 6.6 months, p < 0.0001, log-rank test). The proportion of patients with a tumour response (CR, CRu, PR) was significantly higher (p < 0.0001, Chi-Square test) in the R-CVP group (80.9%) than the CVP group (57.2%). Treatment with R-CVP significantly prolonged the time to disease progression or death compared to CVP, 33.6 months and 14.7 months, respectively (p < 0.0001, log-rank test). The median duration of response was 37.7 months in the R-CVP group and was 13.5 months in the CVP group (p < 0.0001, log-rank test). The difference between the treatment groups with respect to overall survival showed a strong clinical benefit (p = 0.029, log-rank test stratified by center): survival rates at 53 months were 80.9% for patients in the R-CVP group compared to 71.1% for patients in the CVP group.
Results from three other randomized trials using rituximab in combination with chemotherapy regimen other than CVP (CHOP, MCP, CHVP/Interferon α) have also demonstrated significant improvements in response rates, time-dependent parameters as well as in overall survival. Key results from all four studies are summarized in Table 1 as follows. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Maintenance therapy: Previously untreated follicular NHL: In a prospective, open label, international, multicenter, phase III trial 1193 patients previously untreated advanced follicular lymphoma received induction therapy with R-CHOP (n = 881), R-CVP (n = 268) or R-FCM (n = 44), according to the investigators' choice. A total of 1078 patients responded to induction therapy, of which 1018 were randomized to rituximab maintenance therapy (n = 505) or observation (n = 513). The two treatment groups were well balanced with regards to baseline characteristics and disease status. Rituximab maintenance treatment consisted of a single infusion of rituximab at 375 mg/m
2 body surface area given every 2 months until disease progression or for a maximum period of two years.
After a median observation time of 25 months from randomization, maintenance therapy with rituximab resulted in a clinically relevant and statistically significant improvement in the primary endpoint of investigator assessed progression-free survival (PFS) as compared to no maintenance therapy in patients with previously untreated follicular NHL. This improvement in PFS was confirmed by an independent review committee (IRC) (Table 2). Significant benefit from maintenance treatment with rituximab was also seen for the secondary endpoints event-free survival (EFS), time to next anti-lymphoma treatment (TNLT) time to next chemotherapy (TNCT) and overall response rate (ORR) (Table 2).
The updated analysis corresponding to a median observation time of 73 months from randomization confirm the results of the primary analysis (Table 2). (See Table 2.)
Click on icon to see table/diagram/image
Rituximab maintenance treatment provided consistent benefit in all subgroups tested: gender (male, female), age (<60 years, ≥60 years), FLIPI score (1, 2 or 3), induction therapy (R-CHOP, R-CVP or R-FCM) and regardless of the quality of response to induction treatment (CR or PR).
Relapsed/Refractory follicular NHL: In a prospective, open label, international, multicentre, phase III trial, 465 patients with relapsed/refractory follicular NHL were randomised. In a first step to induction therapy with either CHOP (cyclophosphamide, doxorubicin, vincristine, prednisolone; n = 231) or rituximab plus CHOP (R-CHOP, n = 234). The two treatment groups were well balanced with regard to baseline characteristics and disease status. A total of 334 patients achieving a complete or partial remission following induction therapy were randomised in a second step to rituximab maintenance therapy (n=167) or observation (n=167). Rituximab maintenance treatment consisted of a single infusion of rituximab at 375 mg/m
2 body surface area given every 3 months until disease progression or for a maximum period of two years.
The final efficacy analysis included all patients randomized to both parts of the study. After a median observation time of 31 months for patients randomised to the induction phase, R-CHOP significantly improved the outcome of patients with relapsed/refractory follicular NHL when compared to CHOP (Table 3). (See Table 3.)
Click on icon to see table/diagram/image
For patients randomized to the maintenance phase of the trial, the median observation time was 28 months from maintenance randomisation. Maintenance treatment with rituximab led to a clinically relevant and statistically significant improvement in the primary endpoint, PFS, (time from maintenance randomisation to relapse, disease progression or death) when compared to observation alone (p < 0.0001, log-rank test). The median PFS was 42.2 months in the rituximab maintenance arm compared to 14.3 months in the observation arm. Using a cox regression analysis, the risk of experiencing progressive disease or death was reduced by 61% with rituximab maintenance treatment when compared to observation (95% CI; 45% - 72%).
Kaplan-Meier estimated progression-free rates at 12 months were 78% in the rituximab maintenance group versus 57% in the observation group. An analysis of overall survival confirmed the significant benefit of rituximab maintenance over observation (p = 0.0039, log-rank test). Rituximab maintenance treatment reduced the risk of death by 56% (95% CI; 22%-75%).
The median time to new anti-lymphoma treatment was significantly longer with rituximab maintenance treatment than with observation (38.8 months versus 20.1 months, p < 0.0001, log-rank test). The risk of starting a new treatment was reduced by 50% (95% CI; 30% - 64%). In patients achieving a CR/CRu (complete response unconfirmed) as best response during induction treatment, rituximab maintenance treatment significantly prolonged the median disease free survival (DFS) compared to the observation group (53.7 versus 16.5 months, p = 0.0003) log-rank test (Table 4). The risk of relapse in complete responders was reduced by 67% (95% CI; 39% - 82%). (See Table 4.)
Click on icon to see table/diagram/image
The benefit of rituximab maintenance treatment was confirmed in all subgroups analysed, regardless of induction regimen (CHOP or R-CHOP) or quality of response to induction treatment (CR or PR) (Table 2). Rituximab maintenance treatment significantly prolonged median PFS in patients responding to CHOP induction therapy (median PFS 37.5 months versus 11.6 months, p < 0.0001) as well as in those responding to R-CHOP induction (median PFS 51.9 months versus 22.1 months, p = 0.0071). Rituximab maintenance treatment also provided a clinically meaningful benefit in terms of overall survival for both patients responding to CHOP and patients responding to R-CHOP in the induction phase of the study.
Rituximab maintenance treatment provided consistent benefit in all subgroups tested gender, age (≤60 years, >60 years), stage (III, IV), WHO performance status (0 versus > 0), B symptoms (absent, present), bone marrow involvement (no versus yes), IPI (0 - 2 versus 3 - 5), FLIPI score (0 - 1, versus 2 versus 3 - 5), number of extra-nodal sites (0 - 1 versus > 1), number of nodal sites (< 5 versus ≥ 5), number of previous regimens (1 versus 2), best response to prior therapy (CR/PR versus NC/PD), haemoglobin (< 12 g/dL versus ≥12 g/dL), β
2 microglobulin (< 3 mg/L versus ≥ 3 mg/L), LDH (elevated, not elevated) except for the small subgroup of patients with bulky disease.
Adult Diffuse large B-cell non-Hodgkin's lymphoma: In a randomized, open-label trial, a total of 399 previously untreated elderly patients (age 60 to 80 years) with diffuse large B-cell lymphoma received standard CHOP chemotherapy (cyclophosphamide 750 mg/m
2, doxorubicin 50 mg/m
2, vincristine 1.4 mg/m
2 up to a maximum of 2 mg on day 1, and prednisolone 40 mg/m
2/day on days 1 - 5) every 3 weeks for eight cycles, or rituximab 375 mg/m
2 plus CHOP (R-CHOP). Rituximab was administered on the first day of the treatment cycle.
The final efficacy analysis included all randomized patients (197 CHOP, 202 R-CHOP), and had a median follow-up duration of approximately 31 months. The two treatment groups were well balanced in baseline characteristics and disease status. The final analysis confirmed that R-CHOP significantly increased the duration of event-free survival (the primary efficacy parameter, where events were death, relapse or progression of lymphoma, or institution of a new anti-lymphoma treatment) (p = 0.0001). Kaplan Meier estimates of the median duration of event-free survival were 35 months in the R-CHOP arm compared to 13 months in the CHOP arm, representing a risk reduction of 41%. At 24 months, estimates for overall survival were 68.2% in the R-CHOP arm compared to 57.4% in the CHOP arm. A subsequent analysis of the duration of overall survival, carried out with a median follow-up duration of 60 months, confirmed the benefit of R-CHOP over CHOP treatment (p = 0.0071), representing a risk reduction of 32%.
The analysis of all secondary parameters (response rates, progression-free survival, disease-free survival, duration of response) verified the treatment effect of R-CHOP compared to CHOP. The complete response rate after cycle 8 was 76.2% in the R-CHOP group and 62.4% in the CHOP group (p = 0.0028). The risk of disease progression was reduced by 46% and the risk of relapse by 51%.
In all patient subgroups (gender, age, age-adjusted IPI, Ann Arbor stage, ECOG, Beta 2 Microglobulin, LDH, Albumin, B-symptoms, Bulky disease, extranodal sites, bone marrow involvement), the risk ratios for event-free survival and overall survival (R-CHOP compared with CHOP) were less than 0.83 and 0.95; respectively. R-CHOP was associated with improvements in outcome for both high- and low-risk patients according to age-adjusted IPI.
Previously untreated and relapsed/refractory chronic lymphocytic leukaemia: In two open-label randomized trials, a total of 817 previously untreated patients and 552 patients with relapsed/refractory CLL were randomized to receive either FC chemotherapy (fludarabine 25 mg/m
2, cyclophosphamide 250 mg/m
2, days 1-3) every 4 weeks for 6 cycles or rituximab in combination with FC (R-FC). Rituximab was administered at a dosage of 375 mg/m
2 during the first cycle one day prior to chemotherapy and at a dosage of 500 mg/m
2 on day 1 of each subsequent treatment cycle. A total of 810 patients (403 R-FC, 407 FC) the first line study (Table 5 and Table 6 as follows) and 552 patients (276 R-FC, 276 FC) for the relapsed/refractory study (Table 7) were analyzed for efficacy.
In the first line study, after a median observation time of 20.7 months, the median progression-free survival (primary endpoint) was 40 months in the R-FC group and 32 months in the FC group (p < 0.0001, log-rank test) (Table 5). The analysis of overall survival showed an improved survival in favour of the R-FC arm (p = 0.0427, log-rank test). These results were confirmed with longer follow-up: after a median observation time of 48.1 months, the median PFS was 55 months in the R-FC group and 33 months in the FC group (p<0.0001, log-rank test) and overall survival analyses continued to show a significant benefit of R-FC treatment over FC chemotherapy alone (p=0.0319, log-rank test). The benefit in terms of PFS was consistently observed in most patient subgroups analyzed according to disease risk at baseline. (i.e. Binet stages A-C) and was confirmed with longer follow-up (see Table 6). (See Tables 5 and 6.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
In the relapsed/refractory study, the median progression-free survival (primary endpoint) was 30.6 months in the R-FC group and 20.6 months in the FC group (p = 0.0002, log-rank test). The benefit in terms of PFS was observed in almost all patient subgroups analyzed according to disease risk at baseline. A slight but not significant improvement in overall survival was reported in the R-FC compared to the FC arm. (See Table 7.)
Click on icon to see table/diagram/image
Results from other supportive studies using rituximab in combination with other chemotherapy regimens (including CHOP, FCM, PC, PCM, bendamustine and cladribine) for the treatment of CLL patients have also demonstrated high overall response rates with promising PFS rates without adding relevant toxicity to the treatment.
Pediatric population: A multicenter, open-label, randomized study of Lymphome Malin B (LMB) chemotherapy (corticosteroids, vincristine, cyclophosphamide, high-dose methotrexate, cytarabine, doxorubicin, etoposide and triple drug [methotrexate/cytarabine/corticosteroid] intrathecal therapy) alone or in combination with rituximab was conducted in pediatric patients with previously untreated advanced stage CD20 positive DLBCL/BL/BAL/BLL. Advanced stage is defined as Stage III with elevated LDH level ("B-high"), [LDH > twice the institutional upper limit of the adult normal values (> Nx2)] or any stage IV or BAL. Patients were randomized to receive either LMB chemotherapy or six intravenous infusions of rituximab at a dose of 375 mg/m
2 BSA in combination with LMB chemotherapy (two during each of the two induction courses and one during each of the two consolidation courses) as per the LMB scheme. A total of 328 randomized patients were included in the efficacy analyses, of which one patient under 3 years of age received rituximab in combination with LMB chemotherapy.
The two treatment arms, LMB (LMB chemotherapy) and R-LMB (LMB chemotherapy with rituximab), were well balanced with regards to baseline characteristics. Patients had a median age of 7 and 8 years in the LMB arm and R-LMB arm, respectively. Approximately half of patients were in Group B (50.6% in the LMB arm and 49.4% in the R-LMB arm), 39.6% in Group C1 in both arms, and 9.8% and 11.0% were in Group C3 in the LMB and R-LMB arms, respectively. Based on Murphy staging, most patients were either BL stage III (45.7% in the LMB arm and 43.3% in the R-LMB arm) or BAL, CNS negative (21.3% in the LMB arm and 24.4% in the R-LMB arm). Less than half of the patients (45.1% in both arms) had bone marrow involvement, and most patients (72.6% in the LMB arm and 73.2% in the R-LMB arm) had no CNS involvement. The primary efficacy endpoint was EFS, where an event was defined as occurrence of progressive disease, relapse, second malignancy, death from any cause, or non-response as evidenced by detection of viable cells in residue after the second CYVE course, whichever occurs first. The secondary efficacy endpoints were OS and CR (complete remission).
At the pre-specified interim analysis with approximately 1 year of median follow-up, clinically relevant improvement in the primary endpoint of EFS was observed, with 1-year rate estimates of 94.2% (95% CI, 88.5% - 97.2%) in the R-LMB arm vs. 81.5% (95% CI, 73.0% - 87.8%) in the LMB arm, and adjusted Cox HR 0.33 (95% CI, 0.14 - 0.79). Upon IDMC (independent data monitoring committee) recommendation based on this result, the randomization was halted and patients in the LMB arm were allowed to cross over to receive rituximab.
Primary efficacy analyses were performed in 328 randomized patients with median follow-up of 3.1 years. The results are described in Table 8. (See Table 8.)
Click on icon to see table/diagram/image
The primary efficacy analysis showed an EFS benefit of rituximab addition to LMB chemotherapy over LMB chemotherapy alone, with an EFS HR 0.32 (90% CI 0.17 - 0.58) from a Cox regression analysis adjusting for national group, histology, and therapeutic group. While no major differences in numbers of patients achieving CR was observed between the two treatment groups, the benefit of rituximab addition to LMB chemotherapy was also shown in the secondary endpoint of OS, with the OS HR of 0.36 (95% CI, 0.16 - 0.81).
Rheumatoid arthritis: The efficacy of rituximab in rheumatoid arthritis has been demonstrated in three pivotal, phase III, randomized, placebo-controlled, double-blind, multicenter studies. Eligible patients had severe active RA, diagnosed according to the criteria of the American College of Rheumatology (ACR). Rituximab was administered as two IV infusions separated by an interval of 15 days. Each course was preceded by an IV infusion of 100 mg methylprednisolone. All patients received concomitant oral methotrexate. In addition, in Study WA17042, all patients received concomitant oral glucocorticoid on days 2 - 7 and on days 8 to 14 following the first infusion.
The retreatment criteria differed between the studies using one of two approaches; 'Treatment to Remission' whereby patients were treated no more frequently than every 6 months if not in DAS28 remission (i.e. DAS28-ESR≥2.6) and 'Treatment as Needed' strategy ('Treatment PRN'), based on disease activity and/or return of clinical symptoms (swollen and tender joint counts ≥ 8) and treated no sooner than every 16 weeks.
Study WA17042 (REFLEX) included 517 patients that had experienced an inadequate response or intolerance to one or more TNF inhibitor therapies (TNF-IR). The primary endpoint was the proportion of patients who achieved an ACR20 response at week 24. Patients received 2 x 1000 mg rituximab or placebo. Patients were followed beyond week 24 for long term endpoints, including radiographic assessment at 56 weeks. During this time patients could receive further courses of rituximab under an open label extension study protocol. In the open-label protocol patients received further courses based on the 'Treatment PRN' criteria. Study WA17045 (SERENE) included 511 patients that had experienced an inadequate response to methotrexate (MTX-IR) and had not received prior biologic therapy. The primary endpoint was the proportion of patients who achieved an ACR20 response at week 24. Patients received either placebo, 2 x 500 mg or 2 x 1000 mg rituximab infusion. Patients were followed beyond week 24 for long term endpoints and could receive further courses of rituximab based on the 'Treatment to Remission' criteria. An active dose comparison was made at week 48.
Disease Activity Outcomes: In these studies, rituximab (2 x 1000 mg) significantly increased the proportion of patients achieving at least a 20% improvement in ACR score compared with patients treated with methotrexate alone (Table 9). Across all development studies the treatment benefit was similar in patients independent of age, gender, body surface area, race, number of prior treatments or disease status. Patients seropositive for disease related auto-antibodies (RF and/or anti CCP) demonstrated consistently high efficacy compared to MTX alone across studies. Efficacy in seropositive patients was higher than that observed in seronegative patients in whom efficacy was modest.
Clinically and statistically significant improvement was also noted on all individual components of the ACR response (tender and swollen joint counts, patient and physician global assessment, disability index scores (HAQ), pain assessment and CRP (mg/dL)). (See Table 9.)
Click on icon to see table/diagram/image
Patients treated with rituximab had a significantly greater reduction in disease activity score (DAS28) than patients treated with methotrexate alone. A good to moderate EULAR response was achieved by significantly more rituximab treated patients compared to patients treated with methotrexate alone (Table 10). (See Table 10.)
Click on icon to see table/diagram/image
Inhibition of Joint Damage: In studies WA17042 and WA17047 structural joint damage was assessed radiographically and expressed as change in modified Total Sharp Score (TSS) and its components, the erosion score and joint space narrowing score.
In Study WA17042, conducted in TNF-IR patients receiving rituximab in combination with methotrexate demonstrated significantly less radiographic progression at 56 weeks than patients from the methotrexate alone group. A higher proportion of patients receiving rituximab also had no erosive progression over 56 weeks.
Study WA17047, conducted in MTX-naïve patients (755 patients with early rheumatoid arthritis of between 8 weeks to four years duration), assessed the prevention of structural joint damage as its primary objective [see General: Rheumatoid Arthritis Patients (RA) Granulomatosis with Polyangiitis (Wegener's) (GPA) and Microscopic Polyangiitis (MPA) Patients and Pemphigus Vulgaris (PV) Patients under Precautions]. Patients received either placebo, 2 x 500 mg or 2 x 1000 mg rituximab infusion. From week 24 patients could receive further courses of rituximab (or placebo to Week 104) based on the 'Treatment to Remission' criteria. The primary endpoint of change in modified Total Sharp Score (TSS) demonstrated that only treatment with rituximab at a dose of 2 x 1000 mg in combination with methotrexate significantly reduced the rate of progression of joint damage (PJD) at 52 weeks compared with placebo + MTX (Table 11). The reduction in PJD was driven mainly by a significant reduction in the change in Erosion Score.
Inhibition of the rate of progressive joint damage was also observed long term. Radiographic analysis at 2 years in study WA17042 demonstrated significantly reduced progression of structural joint damage in patients receiving rituximab (2 x 1000 mg) + MTX compared to MTX alone as well as a significantly higher proportion of patients with no progression of joint damage over the 2 year period. (See Table 11.)
Click on icon to see table/diagram/image
Quality of Life Outcomes: Rituximab treated patients reported an improvement in all patient-reported outcomes (HAQ-DI, FACIT-Fatigue and SF-36 questionnaires). Significant reductions in disability index (HAQ-DI), fatigue (FACIT-Fatigue), and improvement in the physical health domain of the SF-36 were observed in patients treated with rituximab compared to patients treated with methotrexate alone. (See Tables 12 and 13.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Laboratory Evaluations: In rheumatoid factor (RF) positive patients, marked decreases were observed in rheumatoid factor concentrations following treatment with rituximab in all three studies (range 45 - 64%, Figure 1). (See Figure 1.)
Click on icon to see table/diagram/image
Plasma total immunoglobulin concentrations, total lymphocytes counts, and white cells generally remained within normal limits following rituximab treatment, with the exception of a transient drop in white cells counts over the first four weeks following therapy. Titers of IgG antigen specific antibody to mumps, rubella, varicella, tetanus toxoid, influenza and streptococcus pneumococci remained stable over 24 weeks following exposure to rituximab in rheumatoid arthritis patients.
Effects of rituximab on a variety of biomarkers was evaluated in patients enrolled into a clinical study. This substudy evaluated the impact of a single treatment course of rituximab on levels of biochemical markers, including markers of inflammation (Interleukin 6, C Reactive protein, Serum amyloid type A protein, Protein S100 isotypes A8 and A9), autoantibody (RF and anti-cyclic citrullinated peptide immunoglobulin) production and bone turnover (osteocalcin and procollagen 1 N terminal peptide (P1NP)). Rituximab treatment, whether as monotherapy or in combination with methotrexate or cyclophosphamide reduced the levels of inflammatory markers significantly, relative to methotrexate alone, over the first 24 weeks of follow-up. Levels of markers of bone turnover, osteocalcin and P1NP, increased significantly in the rituximab groups compared to methotrexate alone.
Long-term Efficacy with Multiple Course Therapy: In clinical studies patients were retreated based on either a 'Treatment to Remission' or a 'Treatment PRN' strategy. Repeat courses of rituximab maintained or improved treatment benefit, irrespective of the treatment strategy (Treatment to Remission or Treatment PRN) (Figure 2). However, Treatment to Remission generally provided better responses and tighter control of disease activity as indicated by ACRn, DAS28-ESR and HAQ-DI scores over time. Patients treated PRN also experienced returning disease symptoms between courses, as evidenced by DAS28-ESR scores which were close to pre-treatment levels prior to each course (Table 14). (See Table 14 and Figure 2.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Adult Patients with Granulomatosis with Polyangiitis (Wegener's) (GPA) and Microscopic Polyangiitis (MPA): Adult Induction of Remission (GPA/MPA Study 1): A total of 197 patients with severe, active Granulomatosis with polyangiitis (Wegener's) (GPA) and Microscopic polyangiitis (MPA) were enrolled and treated in an active controlled, randomized, double-blind, multicenter, non-inferiority study. Patients were 15 years of age or older, diagnosed with severely, active Granulomatosis with polyangiitis (Wegener's) (75% of patients) or Microscopic Polyangiitis (MPA) (24% of patients) according to the Chapel Hill Consensus conference criteria (1% of patients had unknown GPA and MPA type). Patients were randomized in a 1:1 ratio to receive either oral cyclophosphamide daily (2 mg/kg/day) for 3 to 6 months, followed by azathioprine or rituximab (375 mg/m
2) once weekly for 4 weeks. Patients in both arms received 1000 mg of pulse intravenous methylprednisolone (or another equivalent-dose glucocorticoid) per day for 1 to 3 days, followed by oral prednisone (1 mg/kg/day, not exceeding 80 mg/day). Prednisone tapering was to be completed by 6 months from the start of study treatment.
The primary outcome measure was achievement of complete remission at 6 months defined as a Birmingham Vasculitis Activity Score for Wegener's Granulomatosis (BVAS/WG) of 0, and off glucocorticoid therapy. The prespecified non-inferiority margin for the treatment difference was 20%. The study demonstrated non-inferiority of rituximab to cyclophosphamide for complete remission at 6 months (Table 15).
Efficacy was observed both for patients with newly diagnosed GPA and MPA and for patients with relapsing disease. (Table 16). (See Tables 15 and 16.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Complete remission at 12 and 18 months: In the rituximab group, 48% of patients achieved CR at 12 months, and 39% of patients achieved CR at 18 months. In patients treated with cyclophosphamide (followed by azathioprine for maintenance of complete remission), 39% of patients achieved CR at 12 months, and 33% of patients achieved CR at 18 months. From month 12 to month 18, 8 relapses were observed in the rituximab group compared with four in the cyclophosphamide group.
Laboratory evaluations: A total of 23/99 (23%) rituximab-treated patients from the induction of remission trial tested positive for ADA by 18 months. None of the 99 rituximab-treated patients were ADA positive at screening. There was no apparent trend or negative impact of the presence of ADA on safety or efficacy in the induction of the remission trial.
Adult Maintenance Treatment (GPA/MPA Study 2): A total of 117 patients (88 with GPA, 24 with MPA, and 5 with renal-limited ANCA-associated vasculitis) in disease remission were randomized to receive azathioprine (59 patients) or rituximab (58 patients) in this prospective, multi-center, controlled, open-label study. Eligible patients were 21 to 75 years of age and had newly diagnosed or relapsing disease in complete remission after combined treatment with glucocorticoids and pulse cyclophosphamide. Patients were ANCA-positive at diagnosis or during the course of their disease; had histologically confirmed necrotizing small-vessel vasculitis with a clinical phenotype of GPA/MPA, or renal limited ANCA-associated vasculitis; or both. Remission-induction therapy included IV prednisone, administered as per the investigator's discretion, preceded in some patients by methylprednisolone pulses, and pulse cyclophosphamide until remission was attained after 4 to 6 months. At that time, and within a maximum of 1 month after the last cyclophosphamide pulse, patients were randomly assigned to receive either rituximab (two 500 mg IV infusion separated by two weeks (on Day 1 and Day 15) followed by 500 mg IV every 6 months for 18 months or azathioprine (administered orally at a dose of 2 mg/kg/day for 12 months, then 1.5 mg/kg/day for 6 months, and finally 1 mg/kg/day for 4 months (treatment discontinuation after these 22 months). Prednisone treatment was tapered and then kept at a low dose (approximately 5 mg per day) for at least 18 months after randomization. Prednisone dose tapering and the decision to stop prednisone treatment after month 18 were left at the investigator's discretion.
All patients were followed until month 28 (10 or 6 months, respectively, after the last rituximab infusion or azathioprine dose).
Pneumocystis jirovecii pneumonia prophylaxis was required for all patients with CD4+ T-lymphocyte counts less than 250 per cubic millimeter.
The primary outcome measure was the rate of major relapse at month 28.
Results: At month 28, major relapse (defined by the reappearance of clinical and/or laboratory signs of vasculitis activity ([BVAS]>0) that could lead to organ failure or damage or could be life threatening) occurred in three patients (5%) in the rituximab group and 17 patients (29%) in the azathioprine group (p=0.0007). Adjusting for the stratification factor using Cox PH modeling, rituximab reduced the risk of major relapse by approximately 86% relative to azathioprine (hazard ratio [HR]: 0.14; 95% confidence interval [CI]: 0.04, 0.47). Minor relapses (not life threatening and not involving major organ damage) occurred in seven patients in the rituximab group (12%) and eight patients in the azathioprine group (14%).
The cumulative incidence rate curves showed that time to first major relapse was longer in patients with rituximab starting from Month 2 and was maintained up to Month 28 (Figure 3). (See Figure 3.)
Click on icon to see table/diagram/image
Laboratory evaluations: A total of 6/34 (18%) of rituximab treated patients from the maintenance therapy clinical trial developed ADA. There was no apparent trend or negative impact of the presence of ADA on safety or efficacy in the maintenance therapy clinical trial.
Pediatric Patients with Granulomatosis with Polyangiitis (Wegener's) (GPA) and Microscopic Polyangiitis (MPA): Study WA25615 (PePRS) was a multicenter, open-label, single-arm, uncontrolled study in 25 patients (≥2 to <18 years old) with active GPA/MPA. The median age of patients in the study was: 14 years (range: 6-17 years) and the majority of patients (20/25 [80%]) were female. A total of 19 patients (76%) had GPA and 6 patients (24%) had MPA at baseline. Eighteen patients (72%) had newly diagnosed disease upon study entry (13 patients with GPA and 5 patients with MPA) and 7 patients had relapsing disease (6 patients with GPA and 1 patient with MPA).
The study design consisted of an initial 6-month remission induction phase, and a minimum 18-month follow-up phase up to a maximum of 54 months (4.5 years). The remission induction regimen consisted of four once weekly IV infusions of rituximab at a dose of 375 mg/m
2 BSA, on study days 1, 8, 15 and 22 in combination with oral prednisolone or prednisone at 1 mg/kg/day (max 60 mg/day) tapered to 0.2 mg/kg/day minimum (max 10 mg/day) by month 6. After the remission induction phase, patients could receive subsequent rituximab infusion on or after month 6 to maintain remission and control disease activity. Patients were to receive a minimum of 3 doses of IV methylprednisolone (30 mg/kg/day, not exceeding 1 g/day) prior to the first rituximab infusion. If clinically indicated, additional daily doses (up to three) of IV methylprednisolone could be given.
All 25 patients completed all four once weekly IV infusions for the 6-month remission induction phase. A total of 24 out of 25 patients completed at least 18 months of follow-up.
The objectives of this study was to evaluate safety, PK parameters, and efficacy of rituximab in pediatric GPA/MPA patients (≥2 to <18 years old). The efficacy objectives of the study were exploratory and principally assessed using the Pediatric Vasculitis Activity Score (PVAS) (Table 17). (See Table 17.)
Click on icon to see table/diagram/image
Cumulative Glucocorticoid dose (IV and Oral) by Month 6: A clinically meaningful decrease in median overall oral glucocorticoid was observed from week 1 (median=45 mg prednisone equivalent dose [IQR: 35-60]) to month 6 (median=7.5 mg [IQR: 4-10]), which was subsequently maintained at month 12 (median - 5 mg [IQR: 2-10]) and month 18 (median=5 mg [IQR: 1-5]).
Follow-Up Treatment: After the 6-month remission induction phase, patients who had not achieved remission or who had progressive disease or flare that could not be controlled by glucocorticoids alone received additional treatment for GPA/MPA, that could include rituximab and/or other therapies, at the discretion of the investigator.
Fourteen out of 25 patients (56%) received additional rituximab treatment at or post month 6, up to month 18. Five patients received four once weekly doses (375 mg/m
2) of rituximab approximately every 6 months; 5 patients received a single dose (375 mg/m
2) of rituximab every 6 months, and a further 4 patients received various other rituximab doses/regimens according to their treating physician. Of the 14 patients, 9 patients achieved PVAS remission by month 6 and sustained remission through month 18: 4 patients achieved remission between month 6 and 12 and sustained remission through month 18. One patient first achieved remission between month 12 and 18.
Laboratory evaluations: A total of 4/25 patients (16%) developed ADA during the overall study period. Limited data shows there was no trend observed in the adverse reactions reported in ADA positive patients.
There was no apparent trend or negative impact of the presence of ADA on safety or efficacy in the pediatric GPA and MPA clinical trials.
Pemphigus Vulgaris: PV Study 1 (Study ML22196): The efficacy and safety of rituximab in combination with short-term low dose glucocorticoid (prednisone) therapy were evaluated in newly diagnosed patients with moderate to severe pemphigus (74 pemphigus vulgaris [PV] and 16 pemphigus foliaceus [PF]) in this randomized, open-label, controlled, multicenter study. Patients were between 19 and 79 years of age and had not received prior therapies for pemphigus. In the PV population, five (13%) patients in the rituximab group and three (8%) patients in the standard prednisone group had moderate disease and 33 (87%) patients in the rituximab group and 33 (92%) in the standard dose prednisone group had severe disease according to disease severity defined by Harman's criteria.
Patients were stratified by baseline disease severity (moderate or severe) and randomized 1:1 to receive either rituximab and low dose prednisone or standard dose prednisone. Patients randomized to the rituximab group received an initial intravenous infusion of 1000 mg rituximab on Study Day 1 in combination with 0.5 mg/kg/day oral prednisone tapered off over 3 months if they had moderate disease or 1 mg/kg/day oral prednisone tapered off over 6 months if they had severe disease, and a second intravenous infusion of 1000 mg on Study Day 15. Maintenance infusions of rituximab 500 mg were administered at months 12 and 18. Patients randomized to the standard dose prednisone group received an initial 1 mg/kg/day oral prednisone tapered off over 12 months if they had moderate disease or 1.5 mg/kg/day oral prednisone tapered off over 18 months if they had severe disease. Patients in the rituximab group who relapsed could receive an additional infusion of rituximab 1000 mg in combination with reintroduced or escalated prednisone dose. Maintenance and relapse infusions were administered no sooner than 16 weeks following the previous infusion.
The primary objective for the study was complete remission (complete epithelialization and absence of new and/or established lesions) at month 24 without the use of prednisone therapy for two months or more (CRoff for ≥2 months). Other efficacy parameters included evaluation of severe and moderate relapses (severity as defined by Harman's criteria and relapse defined as the appearance of ≥3 new lesions a month that did not heal spontaneously within 1 week, or the extension of established lesions in a patient who had achieved disease control), evaluation of the total median cumulative dose of prednisone, and the median duration of complete remission off corticosteroid therapy.
PV study 1 Results: The study demonstrated superiority of rituximab and low dose prednisone over standard dose prednisone in achieving CRoff ≥ 2 months at month 24 in PV patients (see Table 18). Additionally, at month 24, the proportion of PV patients with CRoff ≥ 3 months was higher in the rituximab and low dose prednisone group compared to the standard dose prednisone group (34 patients [90%] vs. 9 patients [25%], p value <0.0001). (See Table 18.)
Click on icon to see table/diagram/image
Rituximab was considered steroid-sparing based on the duration that PV patients were off glucocorticoid therapy and cumulative exposure to glucocorticoids in the rituximab group compared to the standard dose prednisone group.
Duration off Glucocorticoid Therapy: Of PV patients who responded at month 24, the median duration of CRoff ≥ 2 months in the rituximab group was 498.5 days compared to 125 days in the standard dose prednisone group.
Glucocorticoid Exposure: The median (min, max) cumulative prednisone dose at month 24 was 5800 mg (2304, 29303) in the rituximab group compared to 20520 mg (2409, 60565) in the standard dose prednisone group.
Severe or Moderate Relapses: At month 24, 9 (24%) PV patients in the rituximab group experienced at least one severe or moderate relapse vs. 18 (50%) PV patients in the standard dose prednisone group.
PV Study 2 (Study WA29330): In a randomized, double-blind, double-dummy, active-comparator, multicenter study, the efficacy and safety of rituximab compared with mycophenolate mofetil (MMF) were evaluated in patients with moderate-to-severe PV receiving 60-120 mg/day oral prednisone or equivalent (1.0-1.5 mg/kg/day) at study entry and tapered to reach a dose of 60 or 80 mg/day by Day 1. Patients had a confirmed diagnosis of PV within the previous 24 months and evidence of moderate-to-severe disease (defined as a total Pemphigus Disease Area Index, PDAI, activity score of ≥ 15).
One hundred and thirty-five patients were randomized to treatment with rituximab 1000 mg administered on Day 1, Day 15, Week 24 and Week 26 or oral MMF 2 g/day for 52 weeks in combination with 60 or 80 mg oral prednisone with the aim of tapering to 0 mg/day prednisone by Week 24.
The primary efficacy objective for this study was to evaluate at week 52, the efficacy of rituximab compared with MMF in achieving sustained complete remission defined as achieving healing of lesions with no new active lesions (i.e., PDAI activity score of 0) while on 0 mg/day prednisone or equivalent, and maintaining this response for at least 16 consecutive weeks, during the 52-week treatment period.
PV Study 2 Results: The study demonstrated the superiority of rituximab over MMF in combination with a tapering course of oral corticosteroids in achieving CR off corticosteroid ≥ 16 weeks at Week 52 in PV patients (Table 19). The majority of patients in the mITT population were newly diagnosed (74%) and 26% of patients had established disease (duration of illness ≥ 6 months and received prior treatment for PV). (See Table 19.)
Click on icon to see table/diagram/image
The analysis of all secondary parameters (including cumulative oral corticosteroid dose, the total number of disease flares, and change in health-related quality of life, as measured by the Dermatology Life Quality Index) verified the statistically significant results of rituximab compared to MMF. Testing of secondary endpoints were controlled for multiplicity.
Glucocorticoid exposure: The cumulative oral corticosteroid dose was significantly lower in patients treated with rituximab. The median (min, max) cumulative prednisone dose at Week 52 was 2775 mg (450, 22180) in the rituximab group compared to 4005 mg (900, 19920) in the MMF group (p=0.0005).
Disease flare: The total number of disease flares was significantly lower in patients treated with rituximab compared to MMF (6 vs. 44, p<0.0001) and there were fewer patients who had at least one disease flare (8.1% vs. 41.3%).
Laboratory evaluations: By week 52, a total of 20/63 (31.7%) (19 treatment-induced and 1 treatment-enhanced) rituximab-treated PV patients tested positive for ADA. There was no apparent negative impact of the presence of ADA on safety or efficacy in PV Study 2.
IMMUNOGENICITY: As with all therapeutic patients, there is the potential for an immune response in patients treated with rituximab. The data reflects the number of patients whose test results were considered positive for antibodies to rituximab using an enzyme-linked immunosorbent assay (ELISA). Immunogenicity assay results may be influenced by several factors including assay sensitivity and specificity, sample handling, timing of sample collection, concomitant medicinal products and underlying disease. For these reasons, comparison of incidence of antibodies to rituximab with the incidence of antibodies in other studies or to other products may be misleading.
Rheumatoid Arthritis: Approximately 10% of patients with rheumatoid arthritis tested positive for anti-drug antibodies (ADA) in the RA clinical studies. The emergence of ADA was not associated with clinical deterioration or with an increased risk of reactions to subsequent infusions in the majority of patients. The presence of ADA may be associated with worsening of infusion or allergic reactions after the second infusion of subsequent courses, and failure to deplete B cells after receipt of further treatment courses has been observed rarely.
Adult and Pediatric Patients with Granulomatosis with Polyangiitis (Wegener's) (GPA) and Microscopic Polyangiitis (MPA): Twenty-three percent (23/99) of rituximab treated patients from the adult GPA and MPA induction of remission trial and 18% (6/34) of rituximab treated patients in the maintenance therapy clinical trial developed ADA.
In the pediatric clinical trial, a total of 4/25 patients (16%) developed ADA during the overall study period. Limited data shows there was no trend observed in the adverse reactions reported in ADA positive patients. There was no apparent trend or negative impact of the presence of ADA on safety or efficacy in the adult and pediatric GPA and MPA clinical trials.
Pemphigus Vulgaris: By 18 months, a total of 19/34 (56%) (14 treatment-induced and 5 treatment-enhanced) rituximab treated PV patients tested positive for ADA. There was no apparent negative impact of the presence of ADA on safety or efficacy in the PV clinical study.
Pharmacokinetics: Distribution: Non-Hodgkin's Lymphoma: Based on a population pharmacokinetic analysis in 298 NHL patients who received single or multiple infusions of rituximab as a single agent or in combination with CHOP therapy, the typical population estimates of nonspecific clearance (CL
1), specific clearance (CL
2) likely contributed by B cells or tumour burden, and central compartment volume of distribution (V
1) were 0.14 L/day, 0.59 L/day, and 2.7 L, respectively. The estimated median terminal elimination half-life of rituximab was 22 days (range, 6.1 to 52 days). Baseline CD19-positive cell counts and size of measurable tumour lesions contributed to some of the variability in CL
2 of rituximab in data from 161 patients given 375 mg/m
2 as an IV infusion for 4 weekly doses. Patients with higher CD19-positive cell counts or tumour lesions had a higher CL
2. However, a large component of inter-individual variability remained for CL
2 after correction for CD19-positive cell counts and tumour lesion size. V
1 varied by body surface area (BSA) and CHOP therapy. This variability in V
1 (27.1% and 19.0%) contributed by the range in BSA (1.53 to 2.32 m
2) and concurrent CHOP therapy, respectively, were relatively small. Age, gender, race, and WHO performance status had no effect on the pharmacokinetics of rituximab. This analysis suggests that dose adjustment of rituximab with any of the tested covariates is not expected to result in a meaningful reduction in its pharmacokinetic variability.
Rituximab at a dose of 375 mg/m
2 was administered as an IV infusion at weekly intervals for 4 doses to 203 patients with NHL naïve to rituximab. The mean C
max following the fourth infusion was 486 µg/mL (range, 77.5 to 996.6 µg/mL). The peak and trough serum levels of rituximab were inversely correlated with baseline values for the number of circulating CD19-positive B-cells and measures of disease burden. Median steady-state serum levels were higher for responders compared with non-responders. Serum levels were higher in patients with International Working Formulation (IWF) subtypes B, C, and D as compared with those with subtype A. Rituximab was detectable in the serum of patients 3 - 6 months after completion of last treatment.
Rituximab at a dose of 375 mg/m
2 was administered as an IV infusion at weekly intervals for 8 doses to 37 patients with NHL. The mean C
max increased with each successive infusion, spanning from a mean of 243 µg/mL (range, 16 - 582 µg/mL) after the first infusion to 550 µg/mL (range, 171 - 1177 µg/mL) after the eighth infusion.
The pharmacokinetic profile of rituximab when administered as 6 infusions of 375 mg/m
2 in combination with 6 cycles of CHOP chemotherapy was similar to that seen with rituximab alone.
Pediatric DLBCL/BL/BAL/BLL: In the clinical trial studying paediatric DLBCL/BL/BAL/BLL, the PK was studied in a subset of 35 patients aged 3 years and older. The PK was comparable between the two age groups (≥3 to <12 years vs. ≥12 to <18 years). After two rituximab intravenous infusions of 375 mg/m
2 in each of the two induction cycles (cycle 1 and 2) followed by one rituximab intravenous infusion of 375 mg/m
2 in each of the consolidation cycles (cycle 3 and 4) the maximum concentration was highest after the fourth infusion (cycle 2) with a geometric mean of 347 µg/mL followed by lower geometric mean maximum concentrations thereafter (Cycle 4: 247 µg/mL). With this dose regimen, trough levels were sustained (geometric means: 41.8 µg/mL (pre-dose Cycle 2; after 1 cycle), 67.7 µg/mL (pre-dose Cycle 3, after 2 cycles) and 58.5 µg/mL (pre-dose Cycle 4, after 3 cycles)). The median elimination half-life in paediatric patients aged 3 years and older was 26 days.
The PK characteristics of rituximab in paediatric patients with DLBCL/BL/BAL/BLL were similar to what has been observed in adult NHL patients.
No PK data are available in the ≥ 6 months to < 3 years age group, however, population PK prediction supports comparable systemic exposure (AUC, Ctrough) in this age group compared to ≥ 3 years (Table 19). Smaller baseline tumor size is related to higher exposure due to lower time dependent clearance, however, systemic exposures impacted by different tumor sizes remain in the range of exposure that was efficacious and had an acceptable safety profile. (See Table 20.)
Click on icon to see table/diagram/image
Chronic Lymphocytic Leukaemia: Rituximab was administered as an IV infusion at a first-cycle dose of 375 mg/m
2 increased to 500 mg/m
2 each cycle for 5 doses in combination with fludarabine and cyclophosphamide in CLL patients. The mean C
max (N = 15) was 408 µg/mL (range, 97 - 764 µg/mL) after the fifth 500 mg/m
2 infusion.
Rheumatoid Arthritis: Following two intravenous infusions of rituximab at a dose of 1000 mg, two weeks apart, the mean terminal half-life was 20.8 days (range, 8.58 to 35.9 days), mean systemic clearance was 0.23 L/day (range, 0.091 to 0.67 L/day), and mean steady-state distribution volume was 4.6 L (range, 1.7 to 7.51 L). Population pharmacokinetic analysis of the same data gave similar mean values for systemic clearance and half-life, 0.26 L/day and 20.4 days, respectively. Population pharmacokinetic analysis revealed that BSA and gender were the most significant covariates to explain inter individual variability in pharmacokinetic parameters. After adjusting for BSA, male subjects had a larger volume of distribution and a faster clearance than female subjects. The gender-related pharmacokinetic differences are not considered to be clinically relevant and dose adjustment is not required.
The pharmacokinetics of rituximab were assessed following two IV doses of 500 mg and 1000 mg on Days 1 and 15 in four studies. In all these studies, rituximab pharmacokinetics were dose proportional over the limited dose range studied. Mean C
max for serum rituximab following first infusion ranged from 157 to 171 µg/mL for 2 x 500 mg dose and ranged from 298 to 341 μg/mL for 2 x 1000 mg dose. Following second infusion, mean C
max ranged from 183 to 198 μg/mL for the 2 x 500 mg dose and ranged from 355 to 404 μg/mL for the 2 x 1000 mg dose. Mean terminal elimination half-life ranged from 15 to 16.5 days for the 2 x 500 mg dose group and 17 to 21 days for the 2 x 1000 mg dose group. Mean C
max was 16 to 19% higher following second infusion compared to the first infusion for both doses.
The pharmacokinetics of rituximab were assessed following two IV doses of 500 mg and 1000 mg upon re-treatment in the second course. Mean C
max for serum rituximab following first infusion was 170 to 175 μg/mL for 2 x 500 mg dose and 317 to 370 μg/mL for 2 x 1000 mg dose. C
max following second infusion, was 207 μg/mL for the 2 x 500 mg dose and ranged from 377 to 386 μg/mL for the 2 x 1000 mg dose. Mean terminal elimination half-life after the second infusion, following the second course, was 19 days for 2 x 500 mg dose and ranged from 21 to 22 days for the 2 x 1000 mg dose. PK parameters for rituximab were comparable over the two treatment courses.
The pharmacokinetic parameters in the anti-TNF inadequate responder population, following the same dosage regimen (2 x 1000 mg, iv, 2 weeks apart), were similar with a mean maximum serum concentration of 369 μg/mL and a mean terminal half-life of 19.2 days.
Adult and Pediatric Granulomatosis with Polyangiitis (Wegener's) (GPA) and Microscopic Polyangiitis (MPA): The PK parameter in adult and pediatric patients with GPA/MPA receiving 375 mg/m
2 rituximab once weekly for four doses are summarized in Table 21. (See Table 21.)
Click on icon to see table/diagram/image
The PK parameters of rituximab in adult GPA/MPA patients appear similar to what has been observed in RA patients (see Distribution as previously mentioned).
Based on a population pharmacokinetic analysis in pediatric patients with GPA/MPA, the PK parameters of rituximab were similar to those in adults with GPA/MPA, once taking into account the BSA effect on clearance and volume parameters.
Pemphigus vulgaris: The PK parameters in adult PV patients receiving rituximab 1000 mg at Days 1, 15, 168, and 182 are summarized in Table 22. (See Table 22.)
Click on icon to see table/diagram/image
Following the first two rituximab administrations (at day 1 and 15, corresponding to cycle 1), the PK parameters of rituximab in patients with PV were similar to those in patients with GPA/MPA and patients with RA.
Following the last two administrations (at day 168 and 182, corresponding to cycle 2), rituximab clearance decreased while the central volume of distribution remained unchanged.
Elimination: See Distribution as previously mentioned.
Pharmacokinetics in Special Populations: Renal impairment: No pharmacokinetic data are available in patients with renal impairment.
Hepatic impairment: No pharmacokinetic data are available in patients with hepatic impairment.
Pediatrics: The effect of body surface area on the pharmacokinetics of rituximab was assessed in a population pharmacokinetic analysis which included 9 children (≥ 6 years to <12 years) and 16 adolescents (12 to <18 years) with GPA/MPA. BSA was a significant covariate on rituximab pharmacokinetics (see Special Dosage Instructions under Dosage & Administration).


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image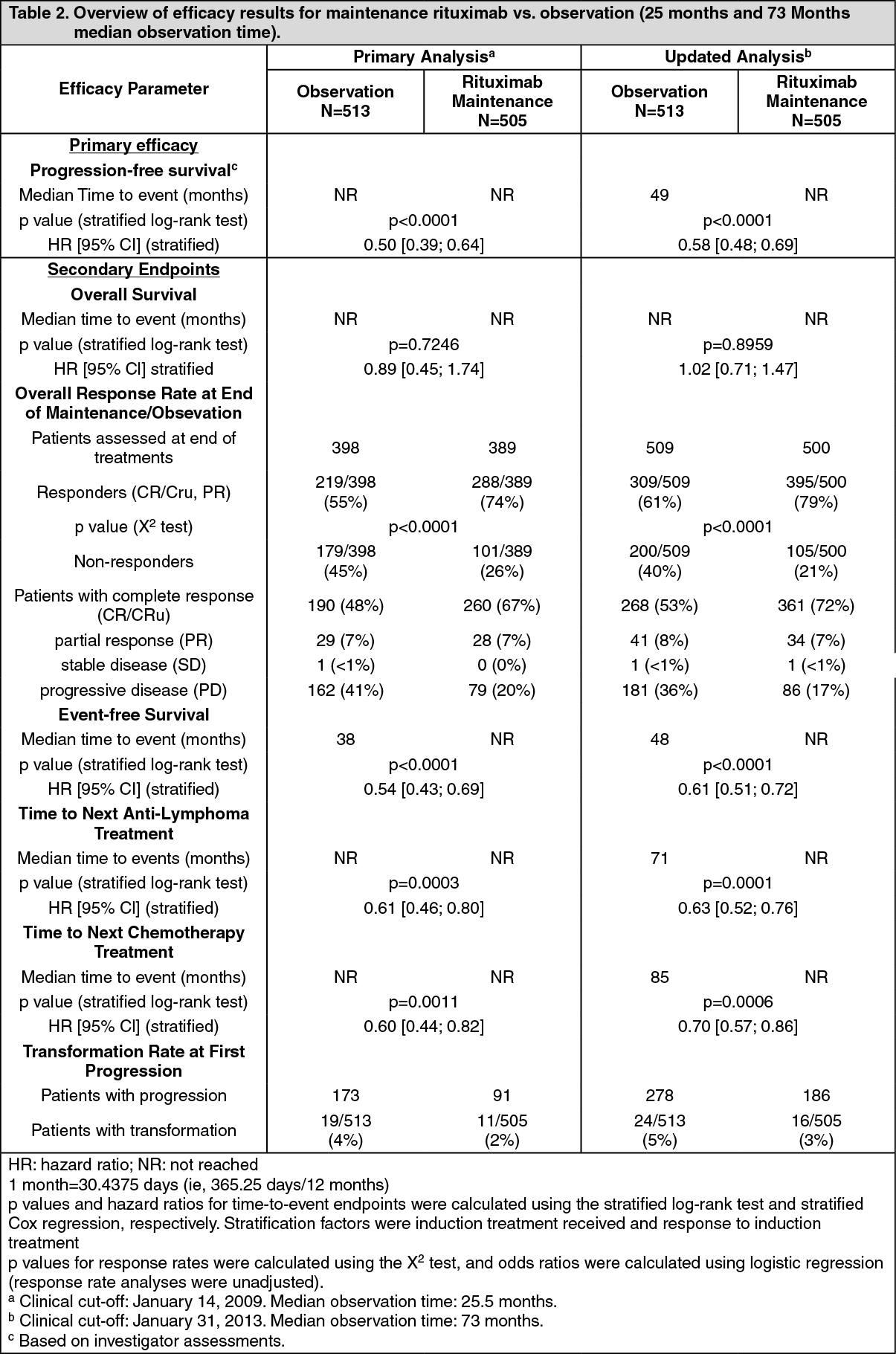 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image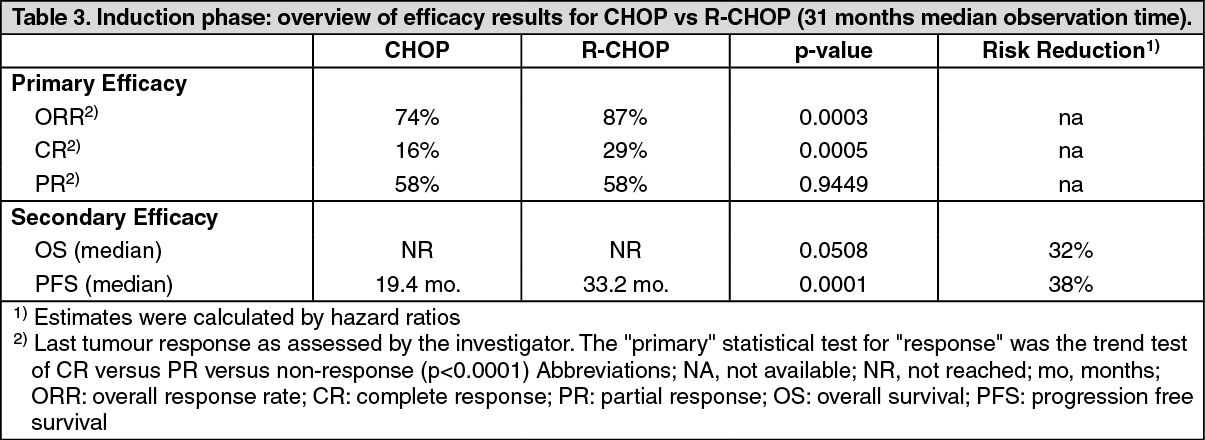 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image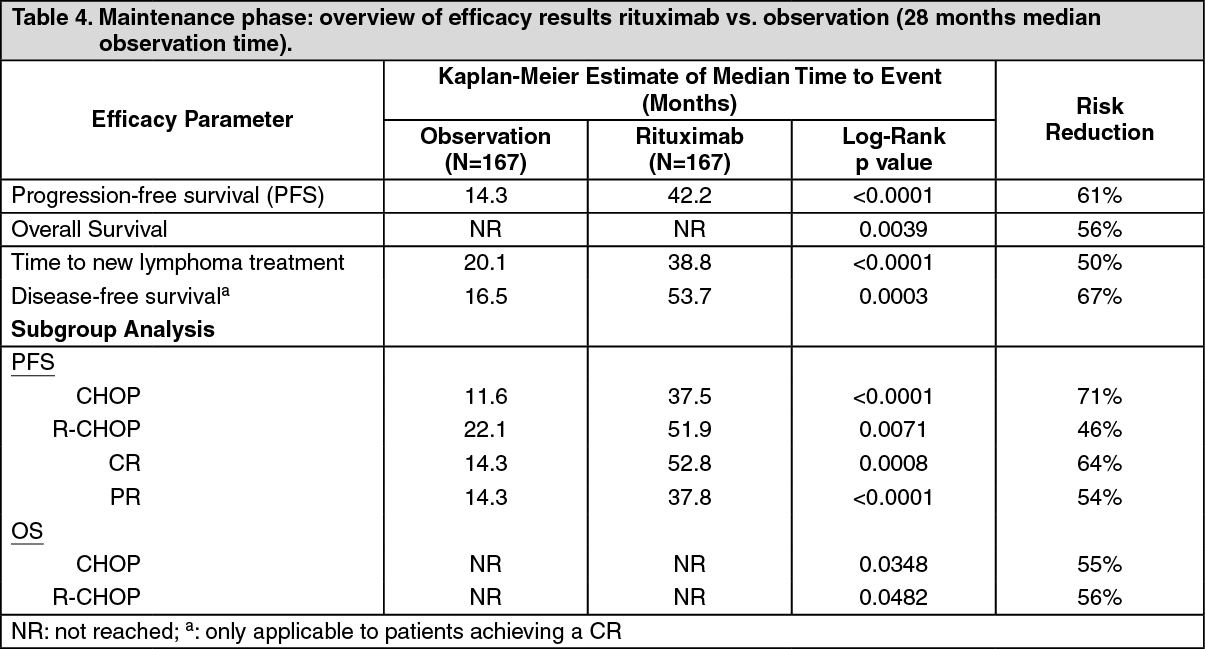 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image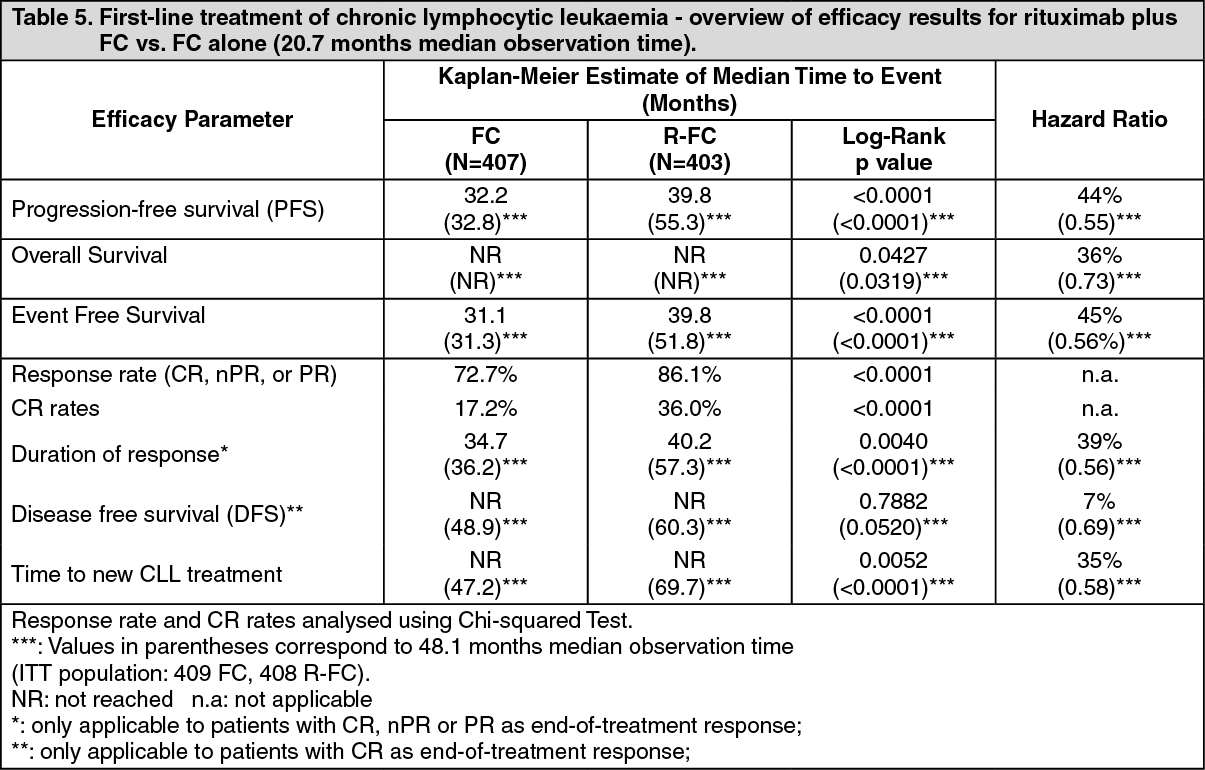 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image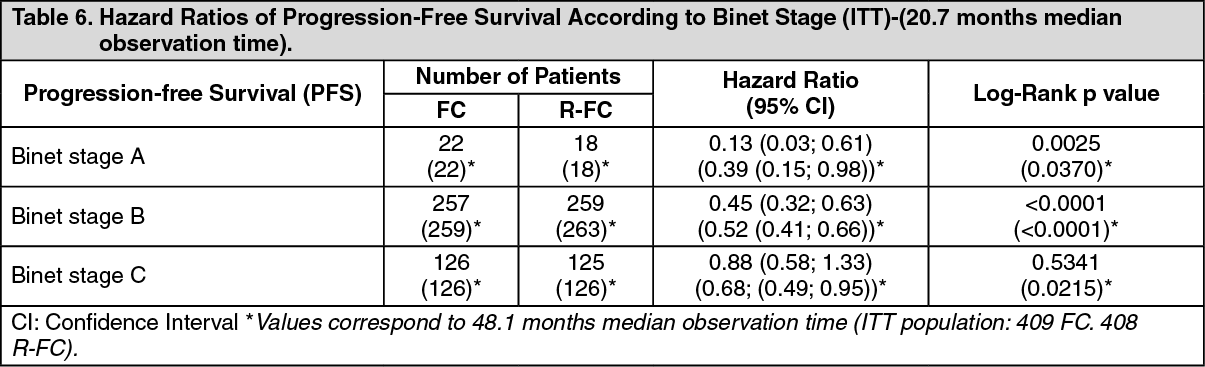 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image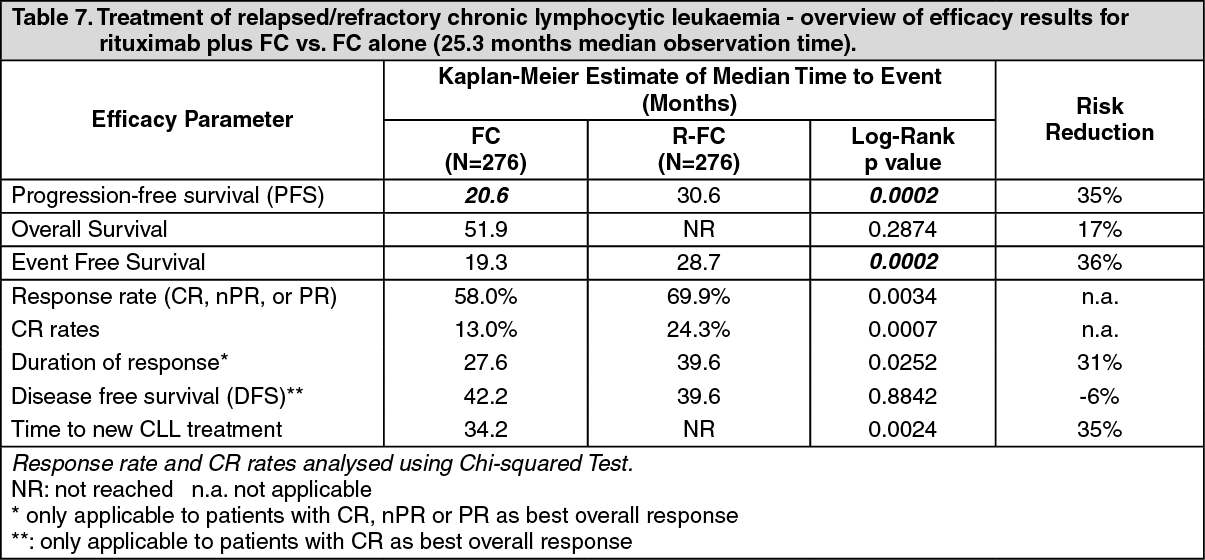 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image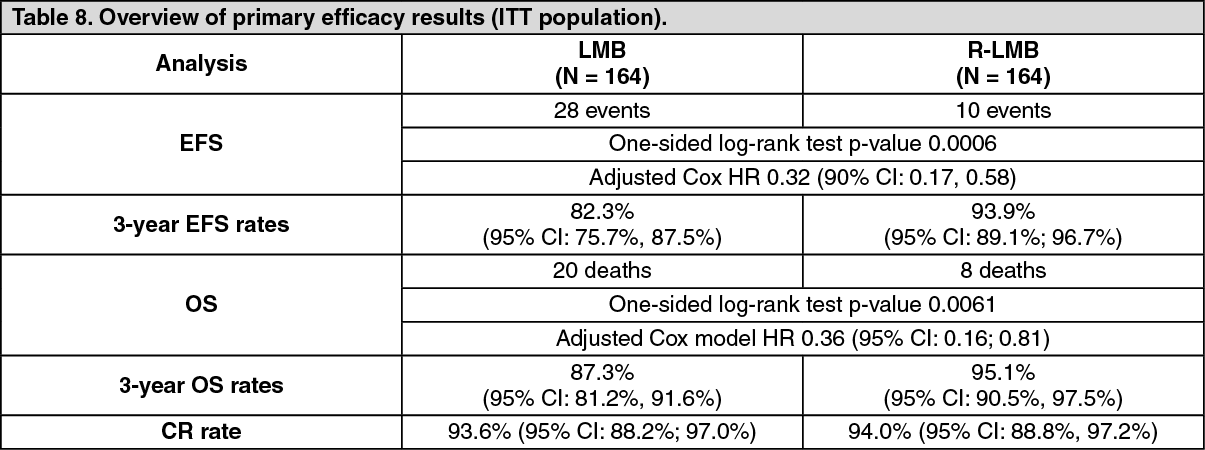 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image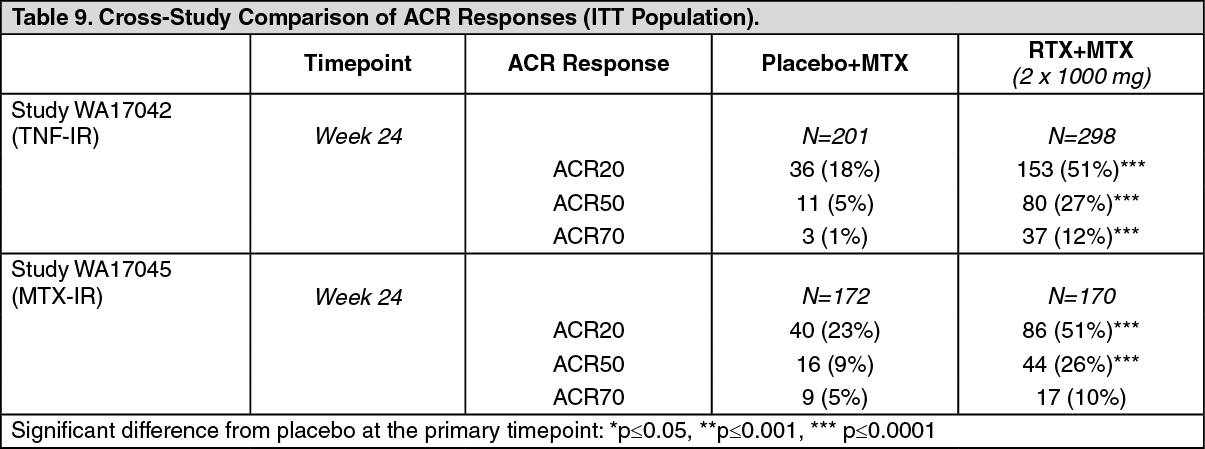 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image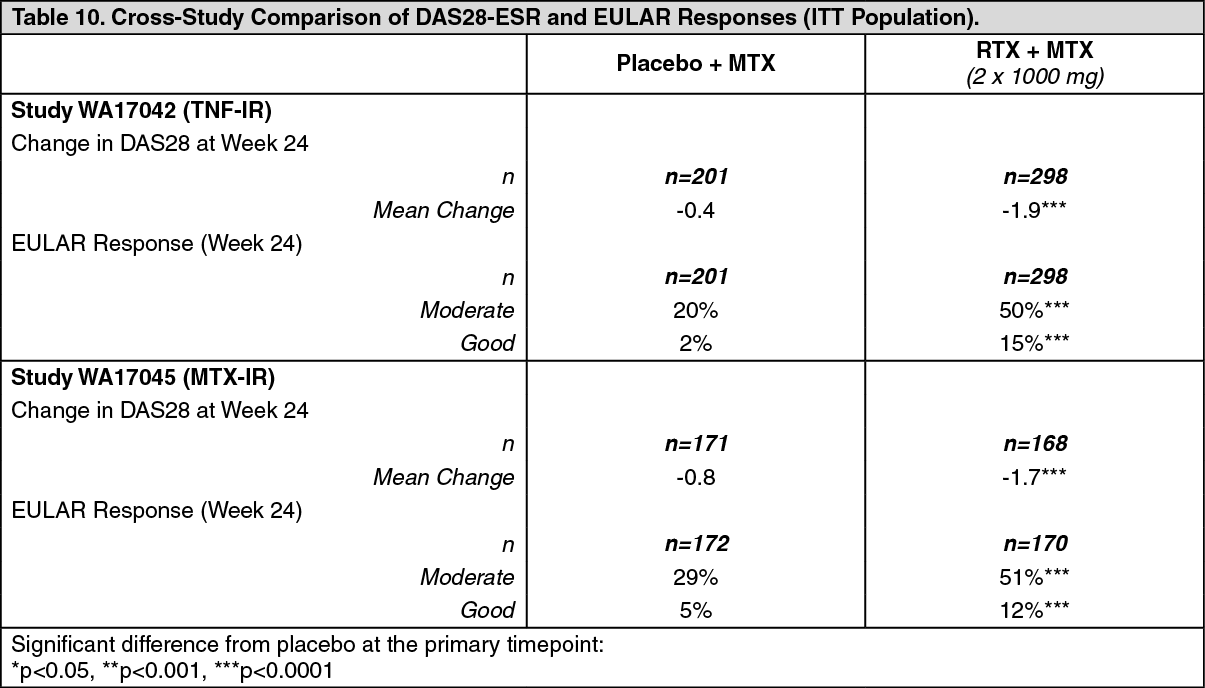 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image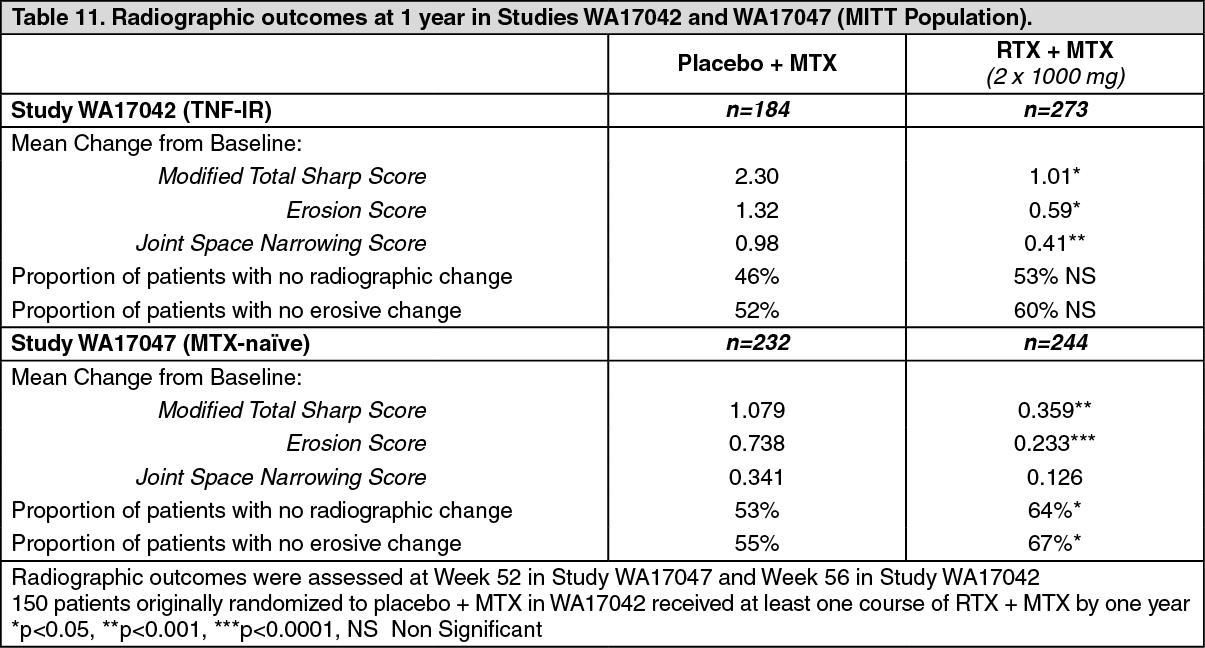 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image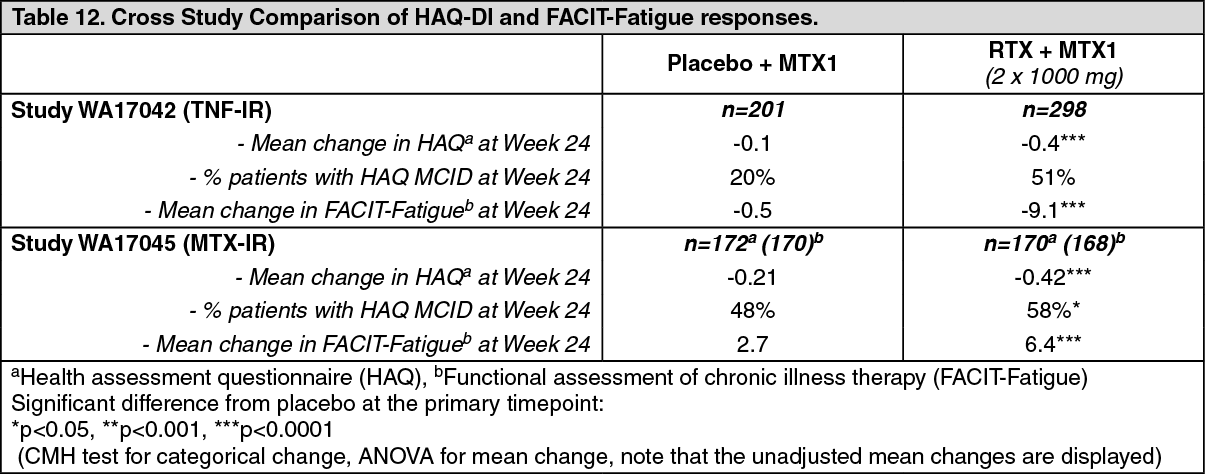 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image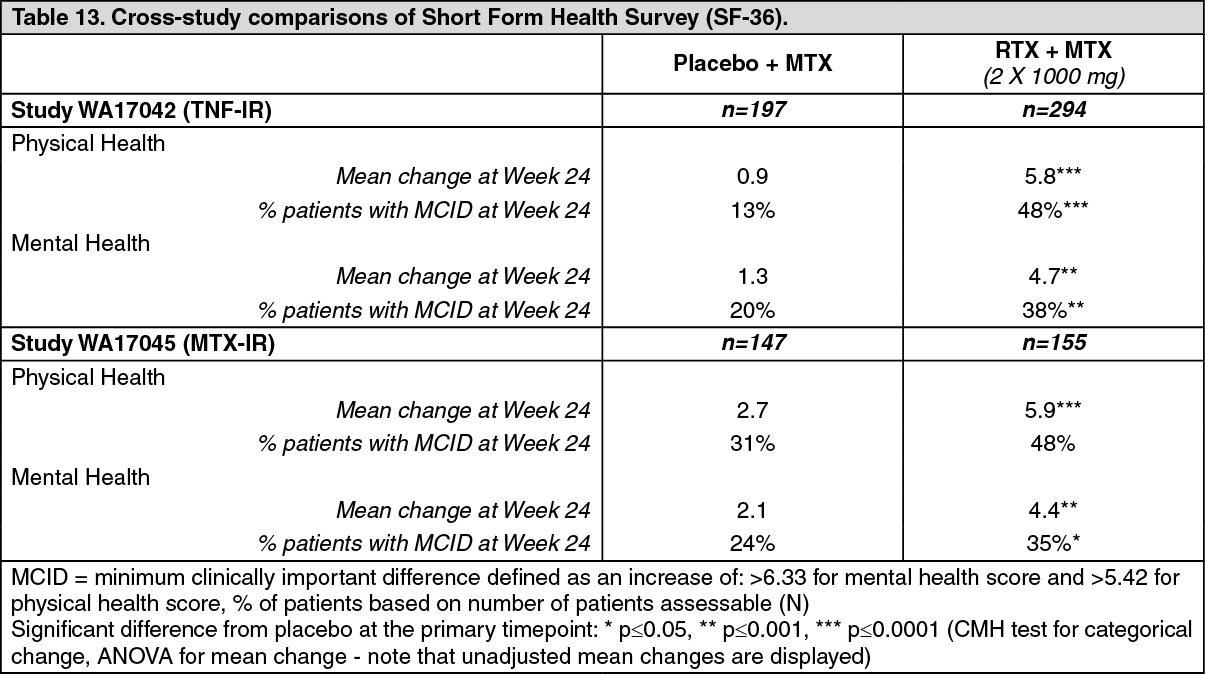 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image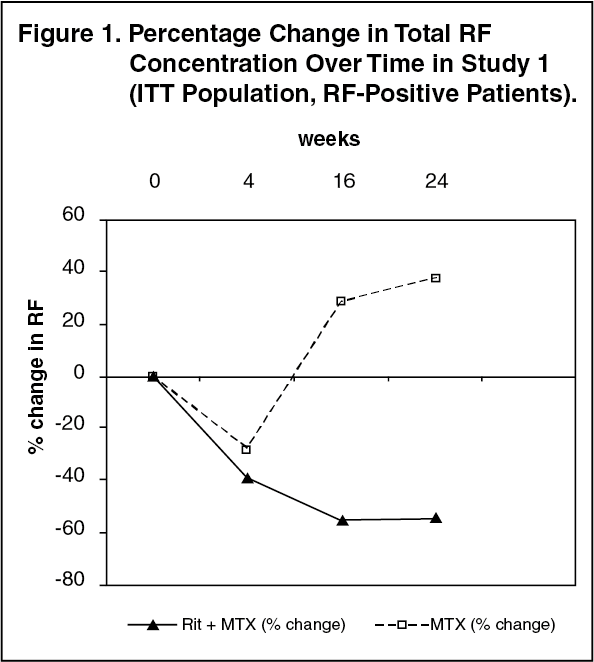 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image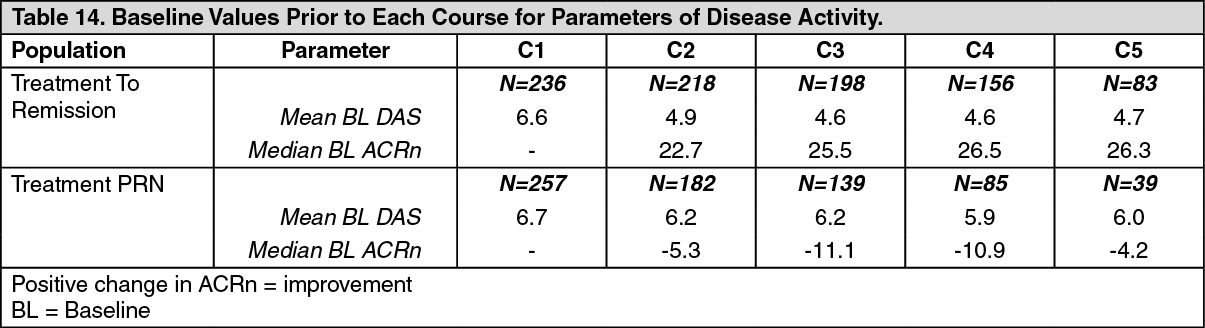 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image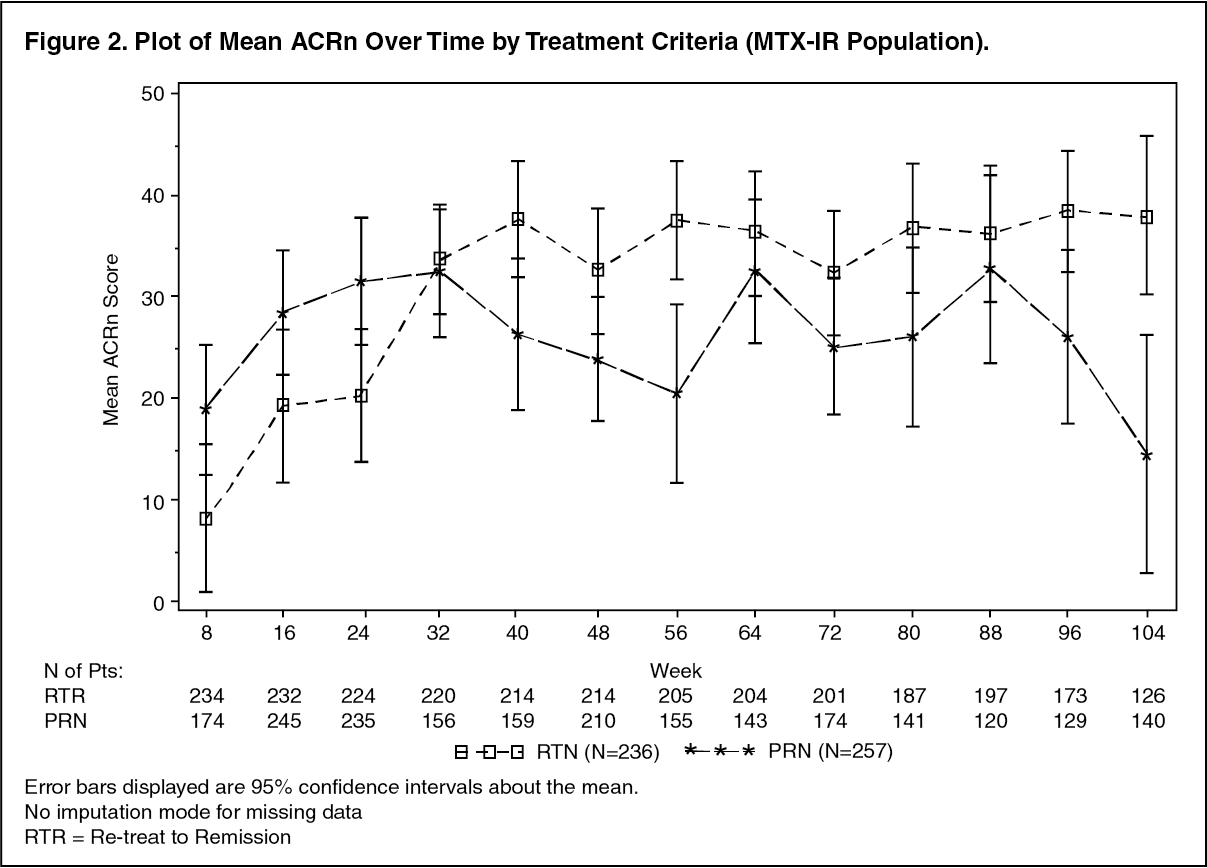 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image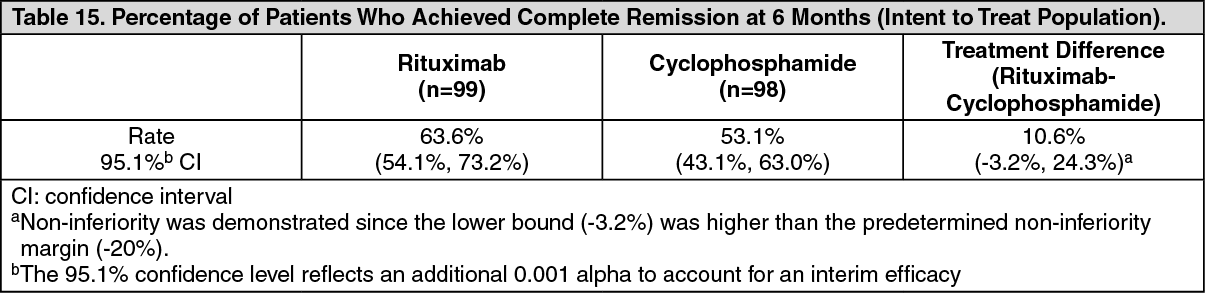 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image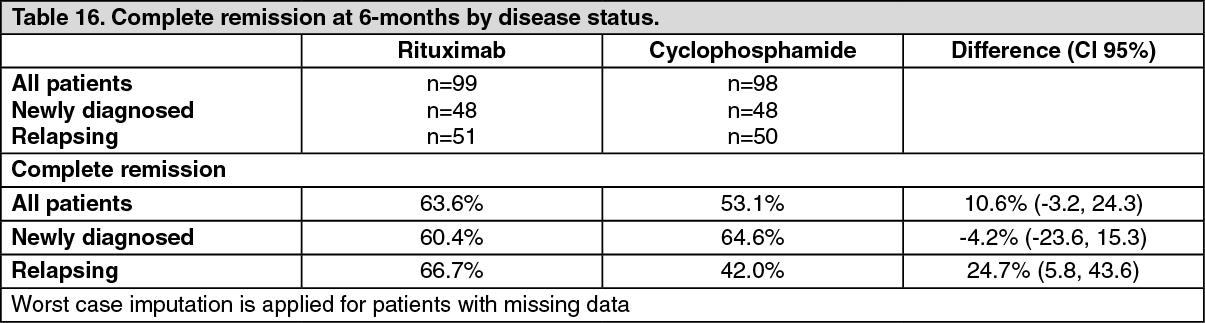 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image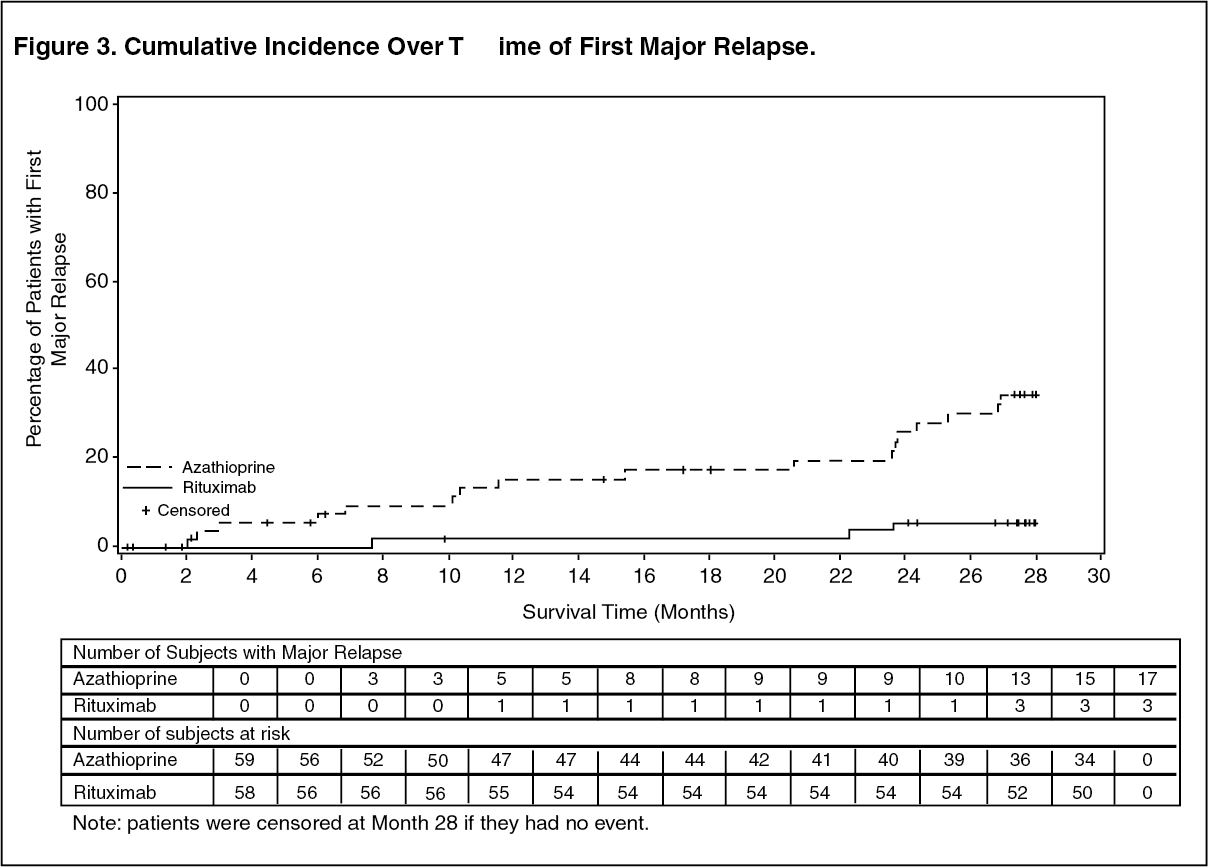 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image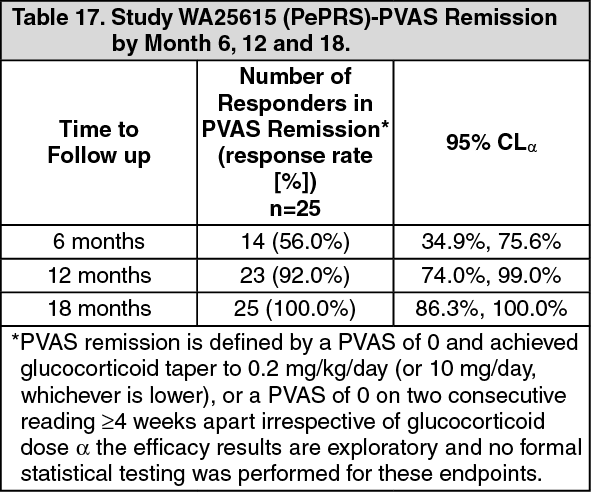 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image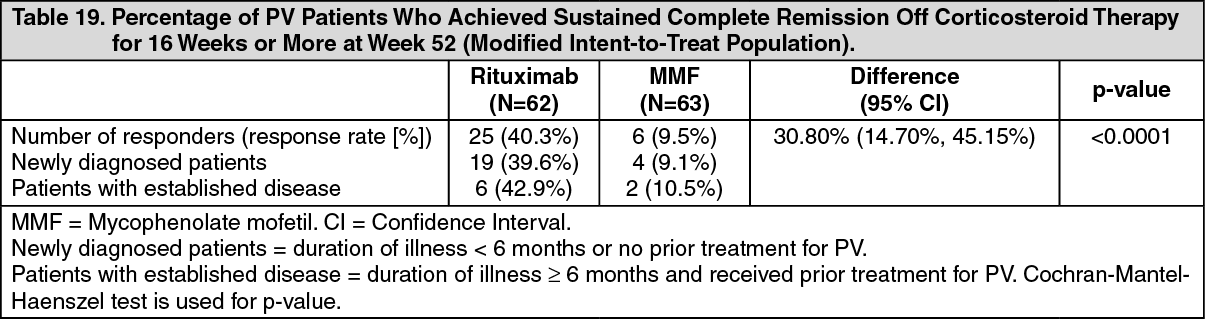 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image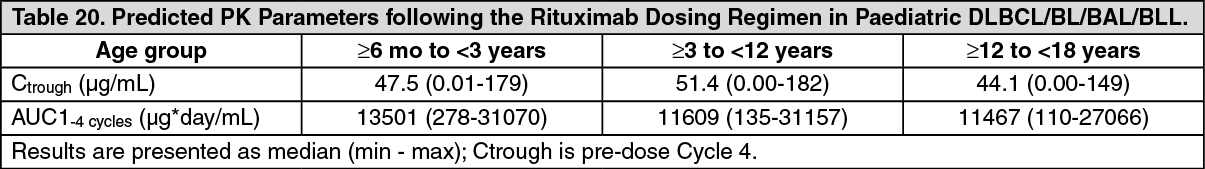 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image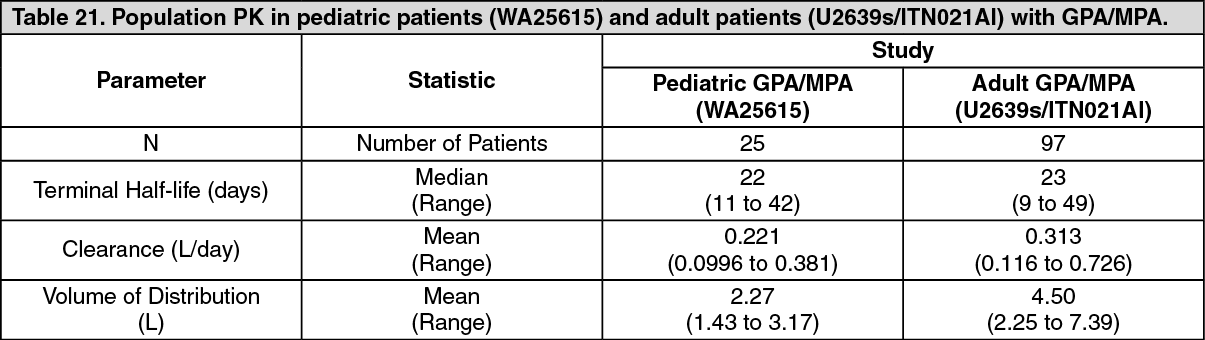 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image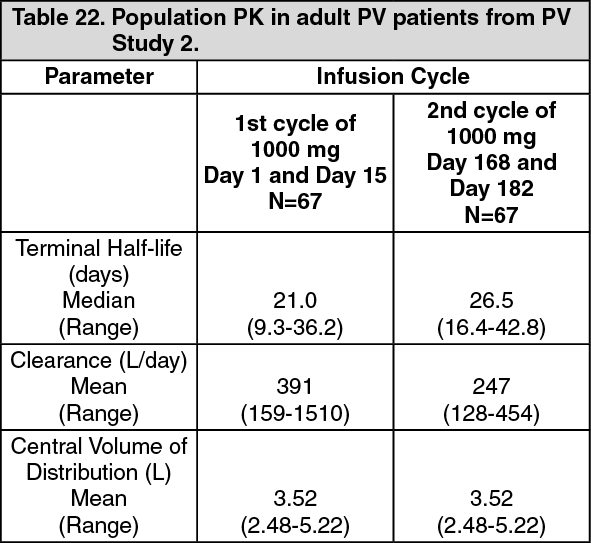 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image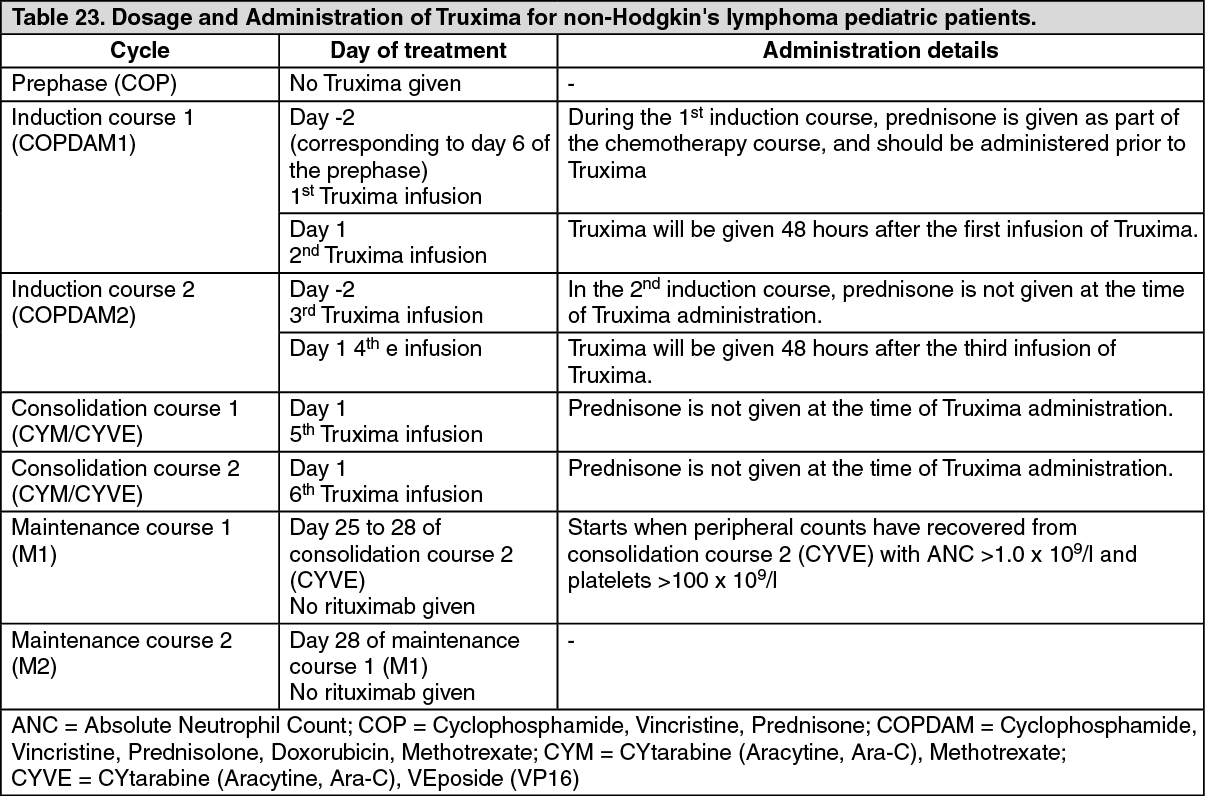 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image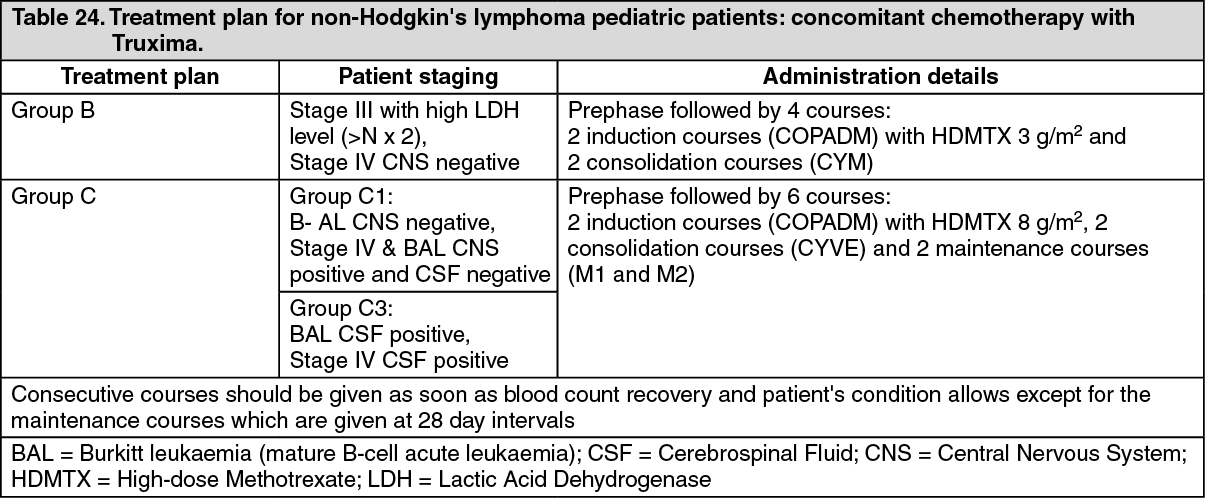 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image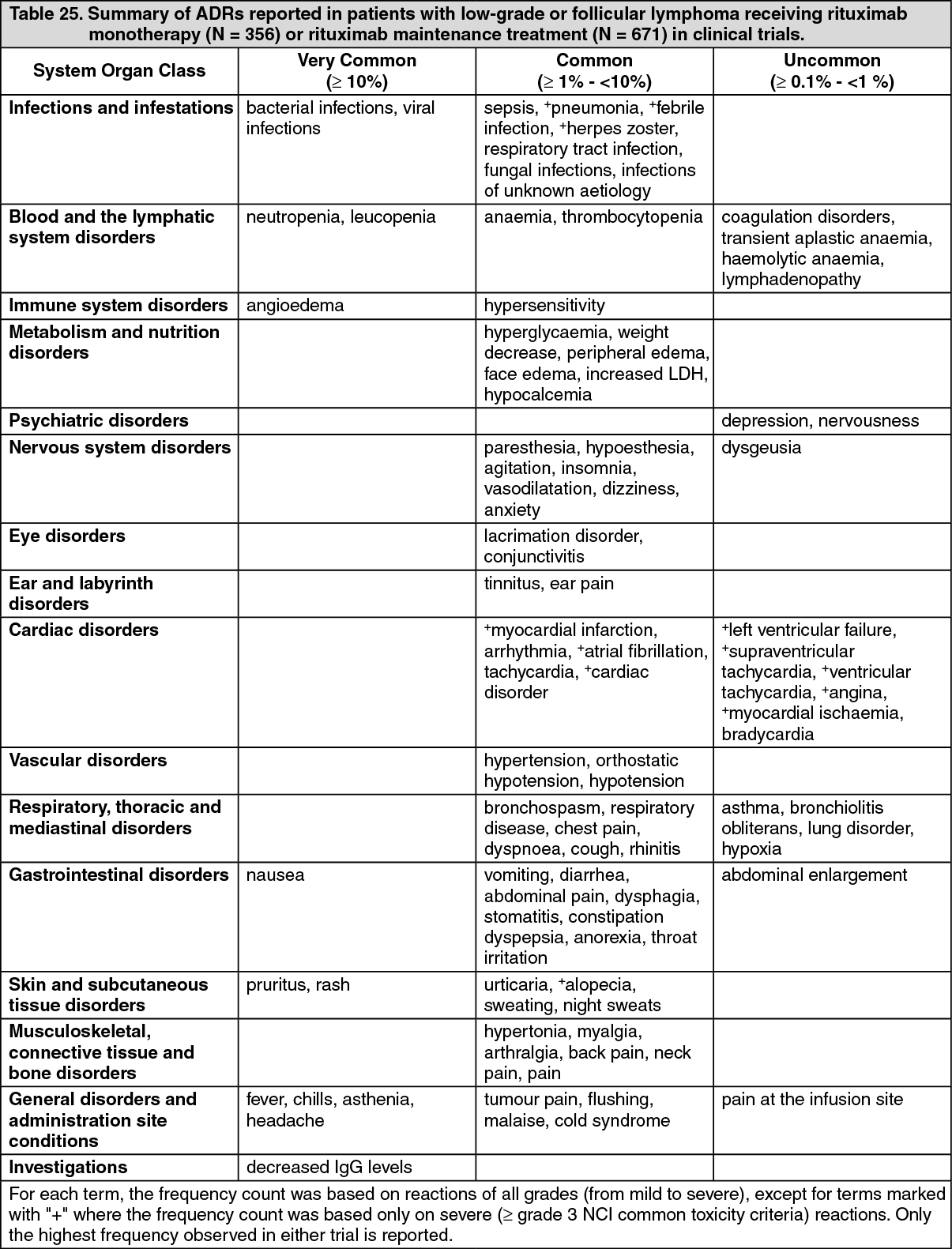 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image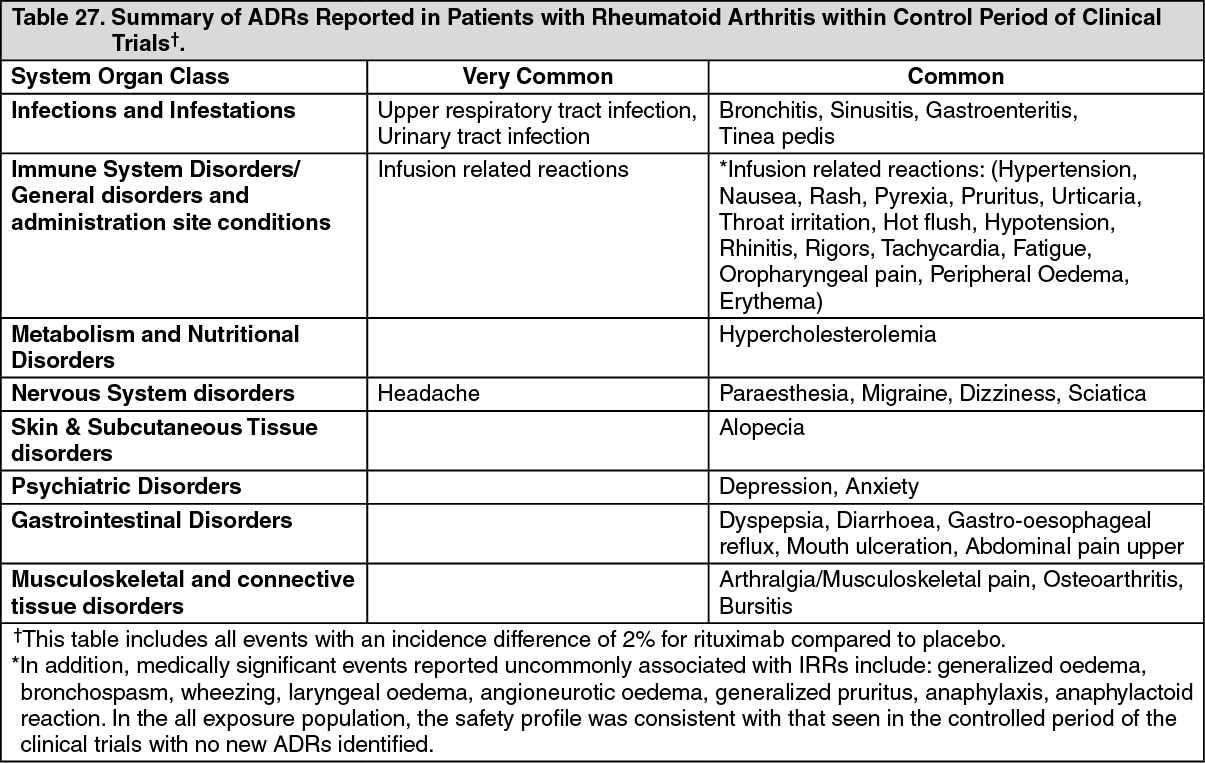 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image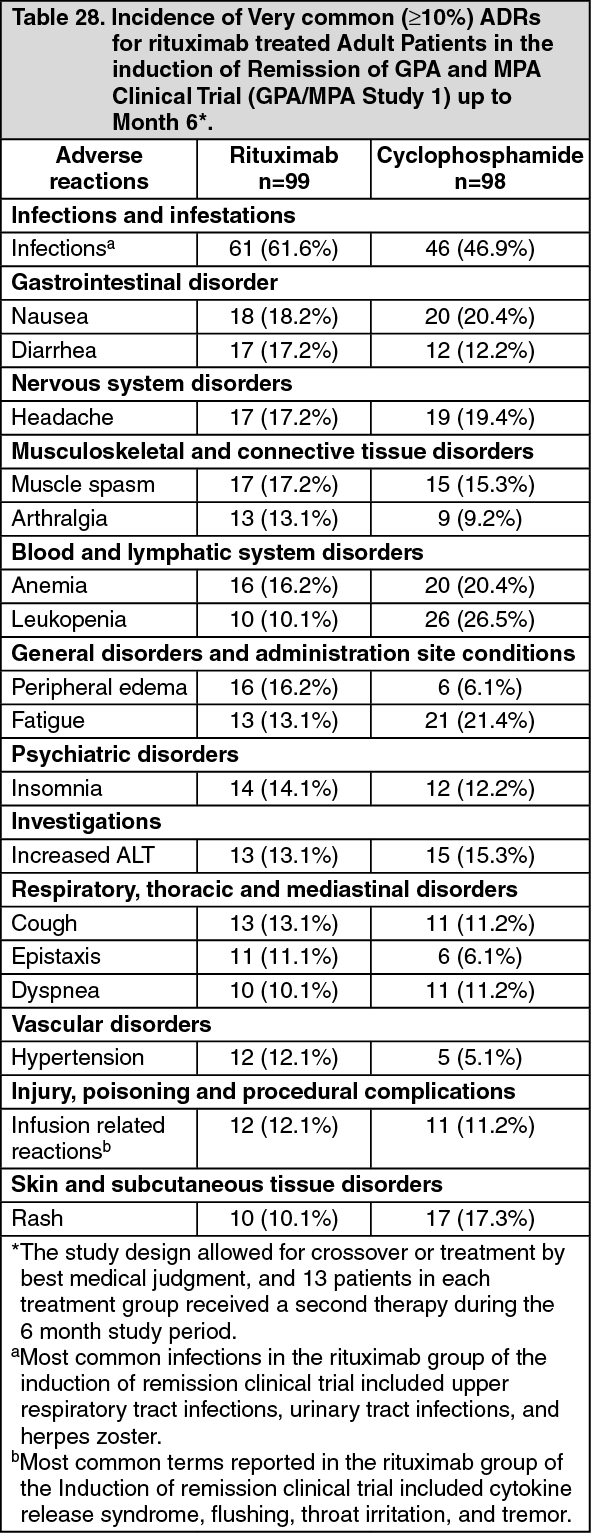 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image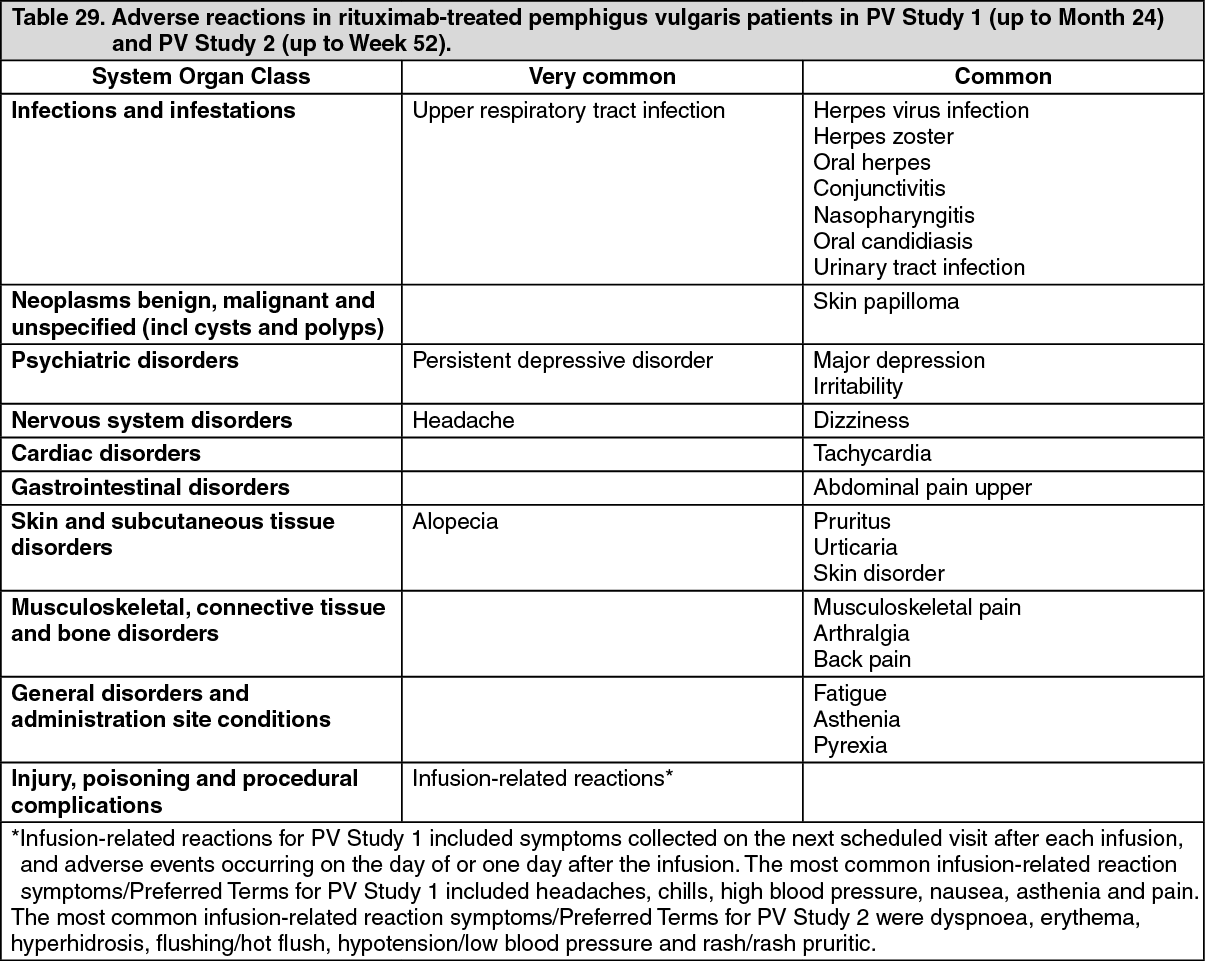 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image


 Sign Out
Sign Out
