Pharmacotherapeutic group: Antineoplastic agents, monoclonal antibodies
and antibody drug conjugates.
ATC code: L01FE02.
PHARMACOLOGY: Pharmacodynamics: Mechanism of action: Panitumumab is a recombinant fully human IgG2 monoclonal antibody that binds with high affinity to the ligand binding domain of human epidermal growth factor receptor (EGFR) and competitively inhibits receptor autophosphorylation induced by all known EGFR ligands. The addition of VECTIBIX to chemotherapy, radiation, or other targeted therapeutic agents in animal studies results in an increase in anti-tumor effects compared to chemotherapy or targeted therapeutic agents alone.
KRAS (Kirsten rat sarcoma 2 viral oncogene homologue) and NRAS (Neuroblastoma RAS viral oncogene homologue) are highly related members of the RAS oncogene family. KRAS and NRAS genes encodes a small, GTP-binding protein involved in signal transduction. A variety of stimuli, including that from the EGFR activates KRAS and NRAS, which in turn stimulates other intracellular proteins to promote cell proliferation, cell survival, and angiogenesis.
Activating mutations in the RAS gene occur frequently in a variety of human tumors and have been implicated in both oncogenesis and tumor progression.
Pharmacodynamic effects: In vitro assays and
in vivo animal studies have shown that panitumumab inhibits the growth and survival of tumor cells expressing EGFR. No anti-tumor effects of panitumumab were observed in human tumor xenografts lacking EGFR expression. The addition of panitumumab to radiation, chemotherapy or other targeted therapeutic agents, in animal studies resulted in an increase in anti-tumor effects compared to radiation, chemotherapy or targeted therapeutic agents alone.
Pharmacokinetics: Panitumumab administered as a single agent or in combination with chemotherapy exhibits nonlinear pharmacokinetics.
Following a single-dose administration of panitumumab as a 1-hour infusion, the area under the concentration-time curve (AUC) increased in a greater than dose-proportional manner and clearance (CL) of panitumumab decreased from 30.6 to 4.6 mL/day/kg as the dose increased from 0.75 to 9 mg/kg. However, at doses above 2 mg/kg, the AUC of panitumumab increases in an approximately dose-proportional manner.
Following the recommended dose regimen (6 mg/kg given once every 2 weeks as a 1-hour infusion), panitumumab concentrations reached steady-state levels by the third infusion with mean (± SD) peak and trough concentrations of 213 ± 59 and 39 ± 14 micrograms/mL, respectively. The mean (± SD) AUC
0-tau and CL were 1,306 ± 374 micrograms·day/mL and
4.9 ± 1.4 mL/kg/day, respectively. The elimination half-life was approximately 7.5 days (range: 3.6 to 10.9 days).
A population pharmacokinetic analysis was performed to explore the potential effects of selected covariates on panitumumab pharmacokinetics. Results suggest that age (21 - 88), gender, race, hepatic function, renal function, chemotherapeutic agents, and EGFR membrane staining intensity (1+, 2+, 3+) in tumor cells had no apparent impact on the pharmacokinetics of panitumumab.
No formal pharmacokinetic studies of panitumumab have been conducted in patients with renal or hepatic impairment.
Clinical studies: Clinical efficacy as monotherapy: The efficacy of VECTIBIX as monotherapy in patients with metastatic colorectal cancer (mCRC) who had disease progression during or after prior chemotherapy was studied in a randomized controlled trial (463 patients, Study 20020408) and open-label, single-arm trials (384 patients, Study 20030167/20030250).
A multinational, randomized, controlled trial was conducted in 463 patients (Study 20020408) with EGFR-expressing metastatic carcinoma of the colon or rectum after confirmed failure of oxaliplatin and irinotecan-containing regimens. Patients were randomized 1:1 to receive VECTIBIX at a dose of 6 mg/kg given once every two weeks plus best supportive care (not including chemotherapy) (BSC) or BSC alone. Patients were treated until disease progression or unacceptable toxicity occurred. Upon disease progression BSC alone patients were eligible to crossover to a companion study and receive VECTIBIX at a dose of 6 mg/kg given once every two weeks.
Of 463 patients, 63% were male. The median age was 62 years (range 27 to 83), and 99% were Caucasian. Three hundred and ninety-six (86%) patients had a baseline ECOG Performance Status of 0 or 1. Sixty-seven percent of patients had colon cancer and 33% had rectal cancer.
The primary endpoint was progression-free survival (PFS). In an analysis adjusting for potential bias from unscheduled assessments, the rate of disease progression or death in patients who received VECTIBIX was reduced by 40% relative to patients that received BSC [hazard ratio = 0.60, (95% CI: 0.49, 0.74), stratified log-rank p < 0.0001]. There was no difference seen in median PFS times as more than 50% of patients progressed in both treatment groups before the first scheduled visit.
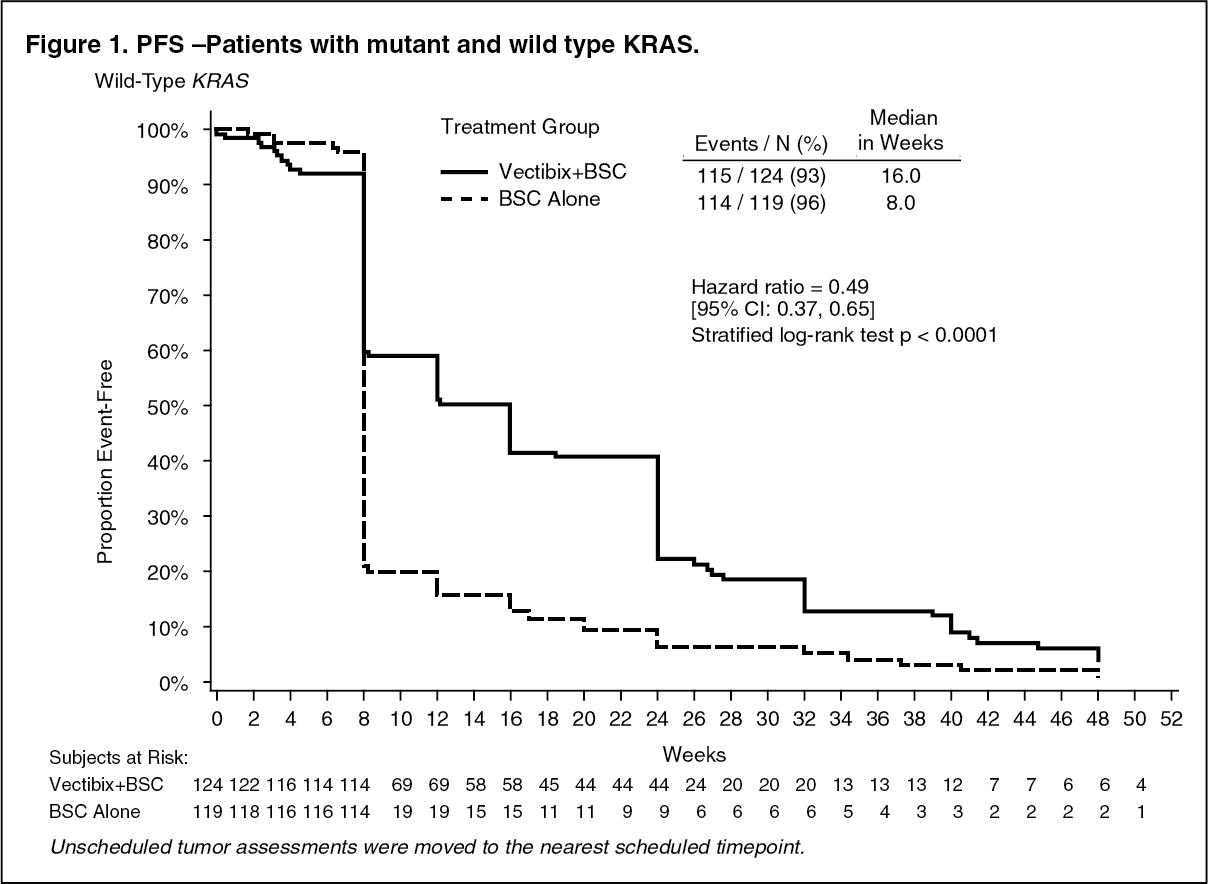
The study was retrospectively analysed by wild-type KRAS status versus mutant KRAS status. KRAS mutation status was determined by analysis of archived paraffin embedded tumor tissue.
Tumor samples obtained from the primary resection of colorectal cancer were analysed for the presence of the seven most common activating mutations in the codon 12 and 13 (Gly12Asp, Gly12Ala, Gly12Val, Gly12Ser, Gly12Arg, Gly12Cys, and Gly13Asp) of the KRAS gene by using an allele-specific polymerase chain reaction. 427 (92%) patients were evaluable for KRAS status of which 184 had mutations. In an analysis adjusting for potential bias from unscheduled assessments the hazard ratio for PFS was 0.49 (95% CI: 0.37 - 0.65) in favor of panitumumab in the wild-type KRAS group and 1.07 (95% CI: 0.77 - 1.48) in the KRAS mutant group. The difference in median PFS in the wild-type KRAS group was 8 weeks. The progression-free survival rates at the first scheduled visit (week 8) in the wild-type KRAS group were 59.7% on VECTIBIX plus BSC and 21.0% on BSC alone, a difference of 38.7% [95% CI: 27.4, 50.0]. The difference in median PFS in the mutant KRAS group was 0 weeks. The progression-free survival rates at the first scheduled visit (week 8) in the mutant KRAS group were 21.4% on VECTIBIX plus BSC and 28.0% on BSC alone, a difference of -6.6% [95% CI: -19.0, 5.9]. There were no differences in overall survival seen in either group. In the wild-type KRAS group the response rate was 17% for panitumumab and 0% for BSC. In the mutant KRAS group there were no responses in either treatment arm. Stable disease rates in the wild-type KRAS group were 34% for panitumumab and 12% for BSC. The stable disease rates in the mutant KRAS group were 12% for panitumumab and 8% for BSC. Response rate (investigator assessment) in patients that crossed over to panitumumab after progression on BSC alone was 22% (95% CI: 14.0, 31.9) for those with wild-type KRAS tumors and 0% (95% CI: 0.0, 4.3) for those with mutant KRAS tumors (Study 20030194). (See Figures 1 and 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Clinical efficacy in combination with chemotherapy: Summary of key efficacy results in pivotal studies: VECTIBIX in combination with chemotherapy. (See Table 1.)
Click on icon to see table/diagram/image
The efficacy of VECTIBIX in combination with oxaliplatin, 5-fluorouracil (5-FU), and leucovorin (FOLFOX) was evaluated in a randomized, controlled trial of 1,183 patients with mCRC with the primary endpoint of progression-free survival (PFS) (Study 20050203). Other key endpoints included the overall survival (OS), objective response rate (ORR), time to response, time to progression (TTP), and duration of response. The study was prospectively analysed by tumor KRAS status. A summary of results in patients with wild-type KRAS mCRC are presented in Table 1.
Primary analysis: The efficacy results in patients with wild-type KRAS (exon 2) mCRC and mutant KRAS mCRC are presented in the table as follows. (See Table 2.)
Click on icon to see table/diagram/image
The results of an exploratory covariate analysis according to ECOG status in patients with wild-type KRAS (exon 2) mCRC are shown as follows: See Table 3.
Click on icon to see table/diagram/image
Final analysis: The efficacy results from the pre-specified final analysis which occurred 2 years after the last patient was enrolled in patients with wild-type KRAS (exon 2) mCRC and mutant KRAS mCRC are presented in the table as follows. (See Table 4.)
Click on icon to see table/diagram/image
The results of an exploratory covariate analysis according to ECOG status in patients with wild-type KRAS (exon 2) mCRC are shown as follows: See Table 5.
Click on icon to see table/diagram/image
Exploratory analysis of overall survival (OS): An exploratory analysis of mature overall survival (> 80% OS events) estimated the treatment effect of panitumumab plus FOLFOX compared with FOLFOX alone on OS by KRAS (exon 2) status. Previous analyses in patients with wild-type KRAS (exon 2) tumor status reported OS with an event rate of 54% of patients in the primary analysis and 68% of patients in the final analysis. Of 656 patients, 535 (82%) with wild-type KRAS (exon 2) mCRC had an OS event at the time of this analysis. Results are shown as follows. (See Table 6.)
Click on icon to see table/diagram/image
Predefined retrospective subset analysis of efficacy and safety by RAS (i.e., KRAS and NRAS) and RAS/BRAF biomarker status: A predefined retrospective subset analysis of 641 patients of the 656 patients with wild-type KRAS (exon 2) mCRC was performed. The primary objective of this analysis was to examine the treatment effect of panitumumab plus FOLFOX compared with FOLFOX alone in patients who were wild-type for RAS (KRAS and NRAS exons 2, 3, and 4) or wild-type for RAS and BRAF (KRAS and NRAS exons 2, 3, and 4 and BRAF exon 15). In this analysis, patient tumor samples with wild-type KRAS exon 2 (codons 12/13) status were tested using Sanger bidirectional sequencing and Surveyor/WAVE analysis in parallel for additional RAS mutations in KRAS exon 3 (codon 61) and exon 4 (codons 117/146) and NRAS exon 2 (codons 12/13), exon 3 (codon 61), and exon 4 (codons 117/146). In the analysis, the incidence of these additional RAS mutations in the wild-type KRAS (exon 2) population was approximately 16%.
In this analysis, BRAF mutation was not found to be predictive of negative outcome for panitumumab treatment.
Results in patients with wild-type RAS mCRC, mutant RAS mCRC and wild-type KRAS (exon 2) mutant RAS mCRC from the primary analysis are presented in the table as follows. (See Table 7.)
Click on icon to see table/diagram/image
Subsequent to the predefined analysis, additional mutations in KRAS and NRAS at exon 3 (codon 59) were identified (n = 7). In an exploratory analysis, adding codon 59 also appeared to be predictive of negative outcomes for panitumumab treatment.
The efficacy of VECTIBIX in combination with irinotecan, 5-fluorouracil (5-FU) and leucovorin (FOLFIRI) was evaluated in a randomized, controlled trial of 1,186 patients with mCRC with the primary endpoints of overall survival (OS) and progression-free survival (PFS) (Study 20050181). Other key endpoints included the objective response rate (ORR), time to response, time to progression (TTP), and duration of response. The study was prospectively analysed by tumor KRAS status. A summary of results in patients with wild-type KRAS mCRC are presented in the previous table.
In patients with wild-type KRAS mCRC (n = 597) a statistically significant difference in PFS in favor of panitumumab was demonstrated (p = 0.0036). The estimated median PFS times were 5.9 months (95% CI: 5.5, 6.7) in the panitumumab plus FOLFIRI arm and 3.9 months (95% CI: 3.7, 5.3) in the FOLFIRI alone arm, an absolute difference of 2.0 months. The hazard ratio was 0.732 (95% CI: 0.593, 0.903), favoring the panitumumab plus FOLFIRI arm. Estimated PFS rate (95% CI) at six (6) months was 49% (42%, 55%) in the panitumumab plus FOLFIRI arm and 35% (29%, 41%) in the FOLFIRI alone arm.
The estimated median OS was 14.5 months (95% CI: 13.0, 16.0) in the panitumumab plus FOLFIRI arm and 12.5 months (95% CI: 11.2, 14.2) in the FOLFIRI alone arm, an absolute difference of 2.0 months. The OS difference did not achieve statistical significance (p = 0.1154). The hazard ratio was 0.854 (95% CI: 0.702, 1.039), favoring the panitumumab plus FOLFIRI arm. Estimated OS rate (95% CI) at twelve (12) months was 59% (53%, 64%) in the panitumumab plus FOLFIRI arm and 53% (47%, 59%) in the FOLFIRI alone arm. Estimated OS rate (95% CI) at eighteen (18) months was 40% (35%, 46%) in the panitumumab plus FOLFIRI arm and 33% (27%, 39%) in the FOLFIRI alone arm. Subsequent chemotherapy (irinotecan, oxaliplatin, or fluoropyrimidine) was given to 142 (47%) subjects in the panitumumab plus FOLFIRI arm and 142 (48%) subjects in the FOLFIRI alone arm. Subsequent anti-EGFR therapy was received by 31 (10%) subjects in the panitumumab plus FOLFIRI arm and 90 (31%) subjects in the FOLFIRI alone arm. The median time to subsequent chemotherapy was 9.9 months in the panitumumab plus FOLFIRI arm and 7.6 months in the FOLFIRI alone arm. The median time to anti-EGFR therapy was 11.8 months (panitumumab plus FOLFIRI) and 7.6 months (FOLFIRI alone). The role of subsequent anti-EGFR therapy or chemotherapy on the estimated OS treatment effect is unknown.
The objective response rate was 35% for patients receiving panitumumab plus FOLFIRI and 10% for patients receiving FOLFIRI alone (all partial responses). The odds ratio for objective response was 5.33 (95% CI: 3.21, 8.60), favoring the panitumumab plus FOLFIRI arm. Stable disease was seen in 116 (39%) patients in the panitumumab plus FOLFIRI arm and 156 (55%) patients in the FOLFIRI alone arm.
The estimated mean (SD) for time to response for responding patients was 2.8 (1.6) months (panitumumab plus FOLFIRI) versus 3.3 (1.4) months (FOLFIRI alone). The duration of response was longer in the panitumumab plus FOLFIRI arm (median 7.6 months [95% CI: 6.7, 9.4]) than in the FOLFIRI alone arm (median 6.6 months [95% CI: 5.7, 10.4]). Time to disease progression was also longer in the panitumumab plus FOLFIRI arm (median 7.3 months [95% CI: 5.9, 7.5]) compared with the FOLFIRI alone arm (median 5.3 months [95% CI: 3.9, 5.7]; hazard ratio 0.683), favoring the panitumumab plus FOLFIRI arm.
Eighteen% (18%) (n = 115) of panitumumab patients had been exposed to prior bevacizumab treatment. PFS and Response Rate were similar regardless of prior bevacizumab treatment.
In an exploratory covariate analysis, longer median OS was observed in the panitumumab plus FOLFIRI arm than in the FOLFIRI alone arm regardless of ECOG performance status (ECOG 0 or 1: 14.7 months vs. 12.8 months, hazard ratio 0.839; 95% CI: 0.685, 1.027; p = 0.0885; ECOG 2: 5.7 months vs. 4.8 months, hazard ratio 1.135; 95% CI: 0.512, 2.517; p = 0.7549).
In patients with mutant KRAS mCRC (n = 486), no significant difference in PFS (HR (95% CI): 0.85 (0.68, 1.06)) and OS (HR (95% CI): 0.94 (0.76, 1.15)) was observed between treatment arms. VECTIBIX is indicated only for the treatment of wild-type KRAS mCRC.
In a randomized, open-label, controlled clinical trial, chemotherapy (oxaliplatin or irinotecan) and bevacizumab were given with and without panitumumab in the first-line treatment of patients with metastatic colorectal cancer (n = 1,053 [n = 823 oxaliplatin cohort, n = 230 irinotecan cohort]) (Study 20040249). Panitumumab treatment was discontinued due to a statistically significant reduction in PFS in patients receiving panitumumab observed in an interim analysis.
The major study objective was comparison of PFS in the oxaliplatin cohort. In the final analysis, the hazard ratio for PFS was 1.27 (95% CI: 1.06, 1.52). Median PFS was 10.0 (95% CI: 8.9, 11.0) and 11.4 (95% CI: 10.5, 11.9) months in the panitumumab and the non-panitumumab arm, respectively. There was an increase in mortality in the panitumumab arm. The hazard ratio for overall survival was 1.43 (95% CI: 1.11, 1.83). Median overall survival was 19.4 (95% CI: 18.4, 20.8) and 24.5 (95% CI: 20.4, 24.5) in the panitumumab arm and the non-panitumumab arm.
An additional analysis of efficacy data by KRAS status did not identify a subset of patients who benefited from panitumumab in combination with oxaliplatin- or irinotecan-based chemotherapy and bevacizumab. For the wild-type KRAS subset of the oxaliplatin cohort, the hazard ratio for PFS was 1.36 with 95% CI: 1.04 - 1.77. For the mutant KRAS subset, the hazard ratio for PFS was 1.25 with 95% CI: 0.91 - 1.71. A trend for OS favoring the control arm was observed in the wild-type KRAS subset of the oxaliplatin cohort (hazard ratio = 1.89; 95% CI: 1.30, 2.75). A trend towards worse survival was also observed with panitumumab in the irinotecan cohort regardless of KRAS mutational status. Overall, panitumumab treatment combined with chemotherapy and bevacizumab is associated with an unfavorable benefit-to-risk profile irrespective of tumor KRAS mutational status.
Toxicology: Preclinical safety data/Nonclinical toxicology: Carcinogenesis: The carcinogenic potential of VECTIBIX has not been evaluated.
Mutagenesis: The mutagenic potential of VECTIBIX has not been evaluated
in vitro or
in vivo.
Impairment of fertility: Formal male fertility studies have not been conducted; however, microscopic evaluation of male reproductive organs from cynomolgus monkeys administered VECTIBIX for 26 weeks at doses ranging up to 5-fold the human dose revealed no differences compared to control male monkeys. Fertility studies conducted in female cynomolgus monkeys showed that VECTIBIX may produce secondary effects that could impact the ability of a woman to become pregnant while receiving VECTIBIX.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image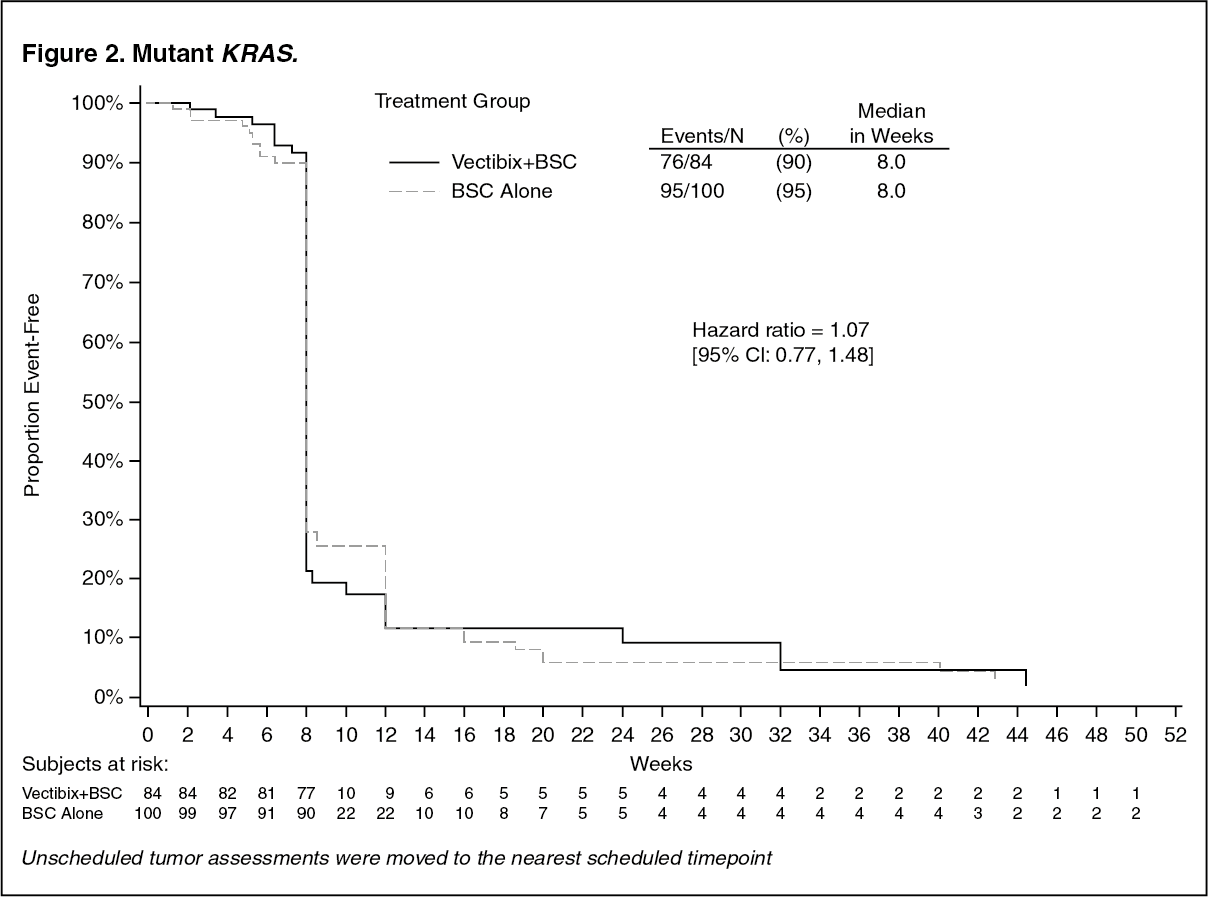 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image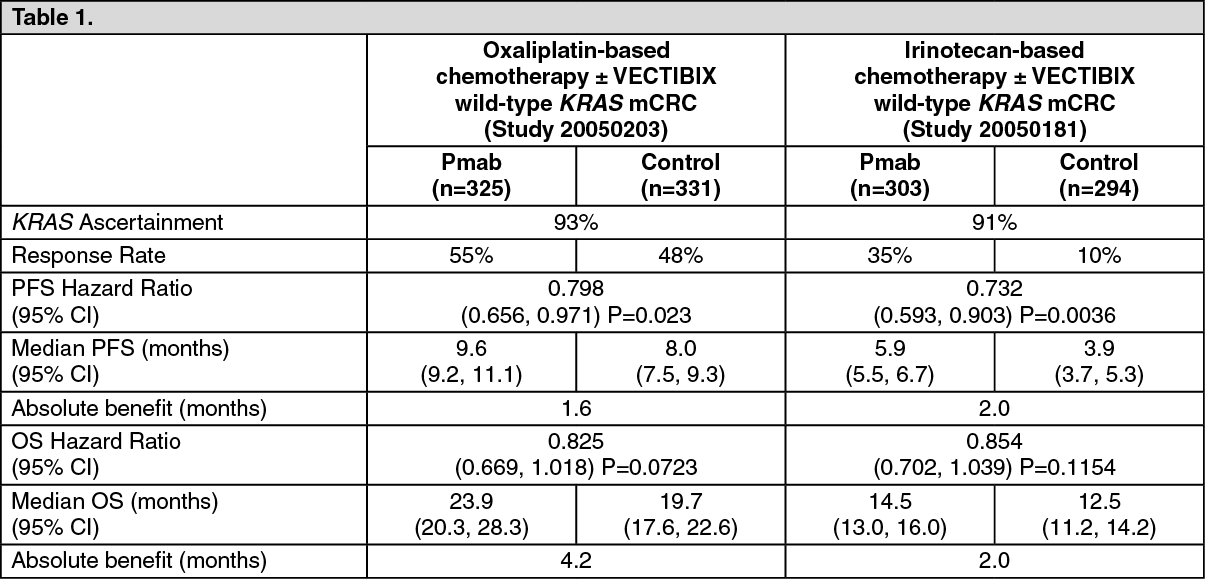 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image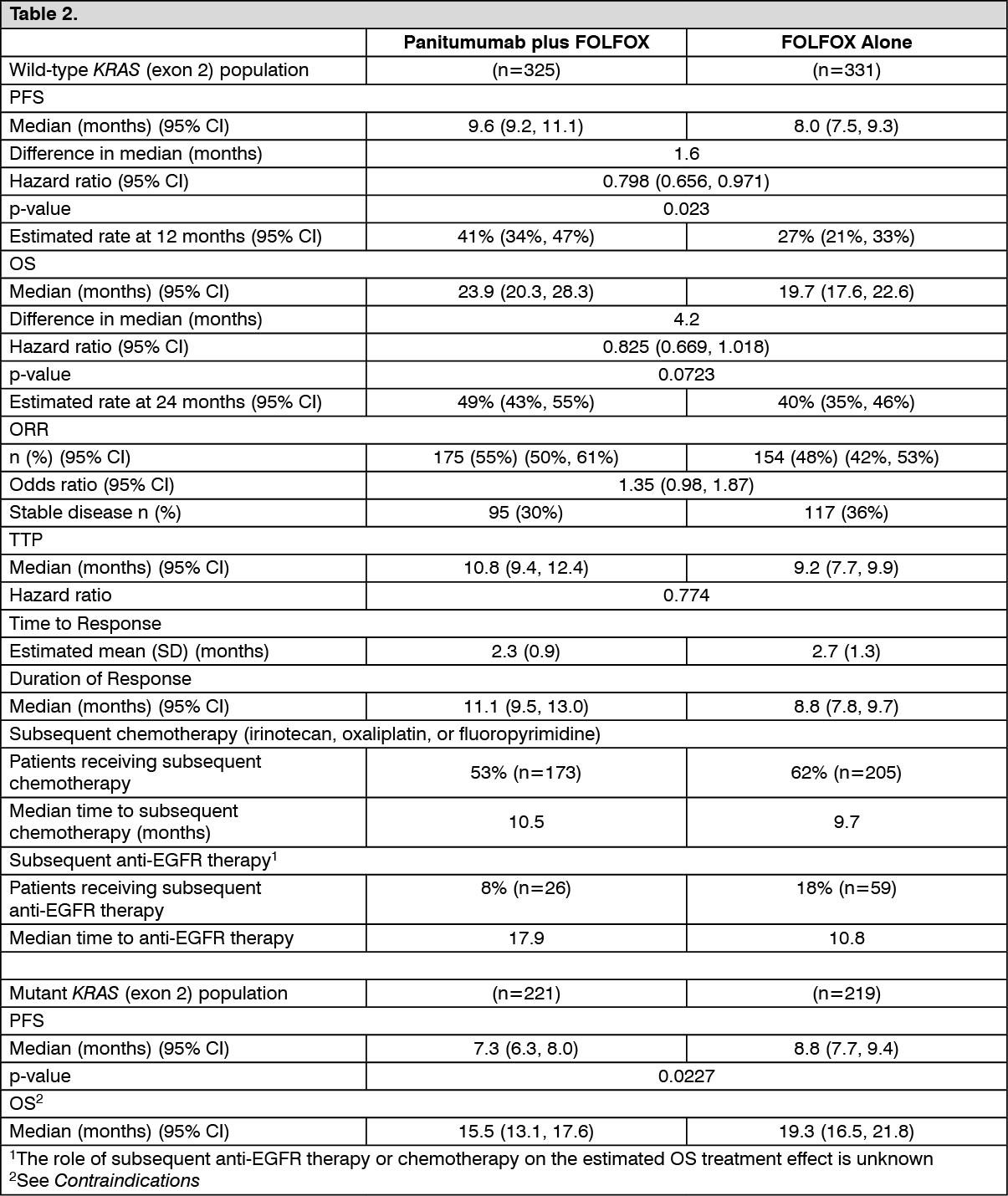 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image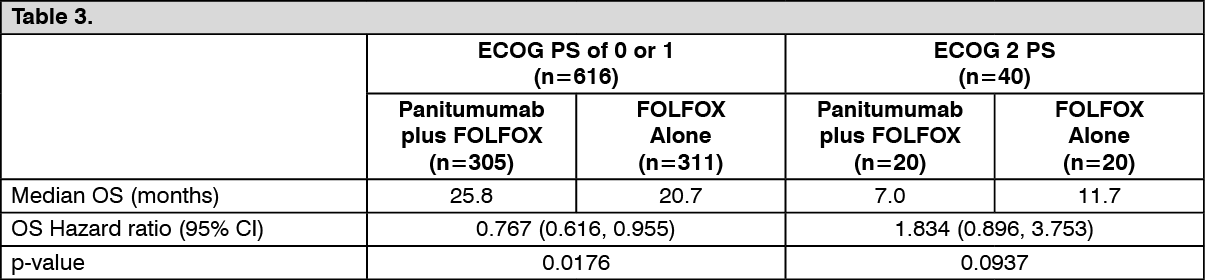 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image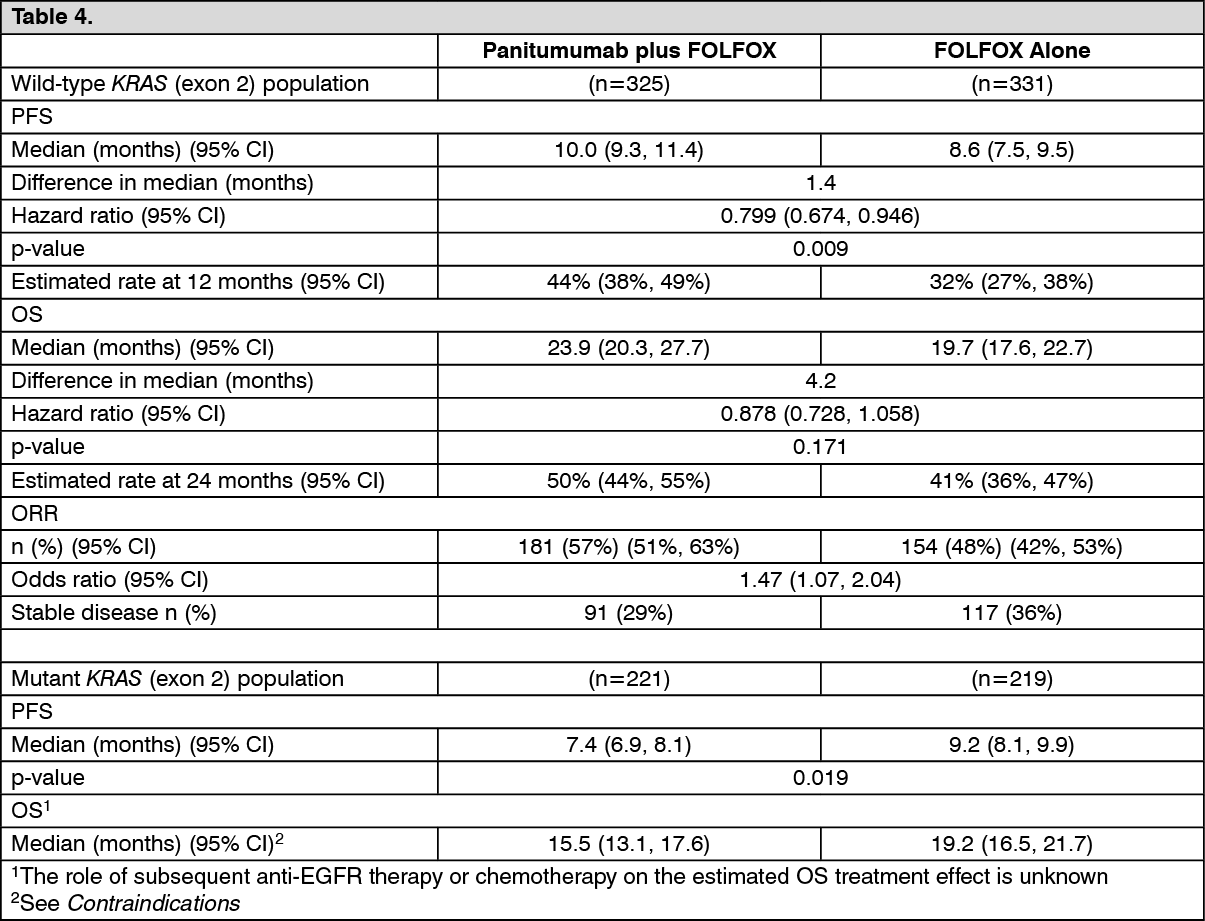 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image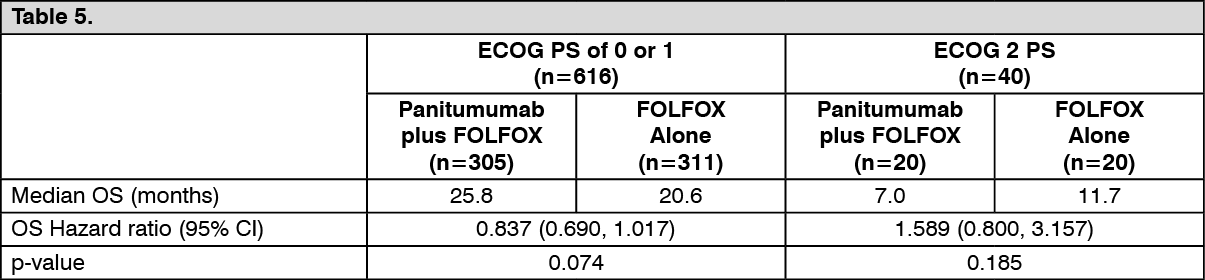 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image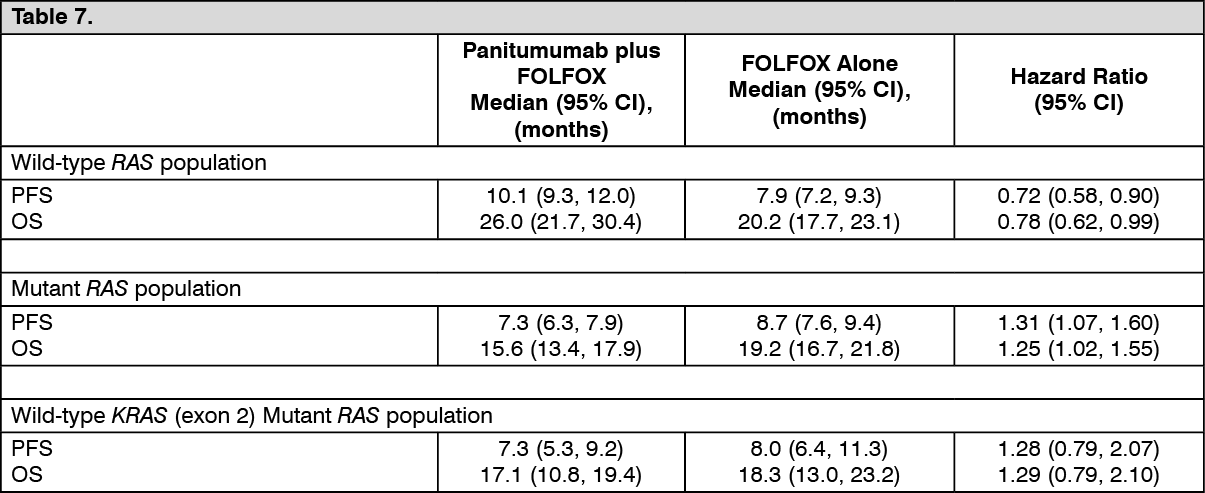 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image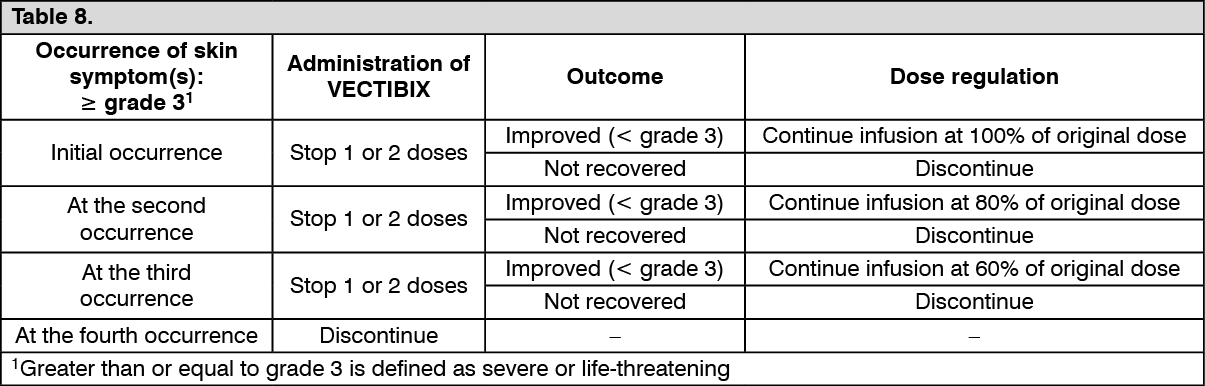 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image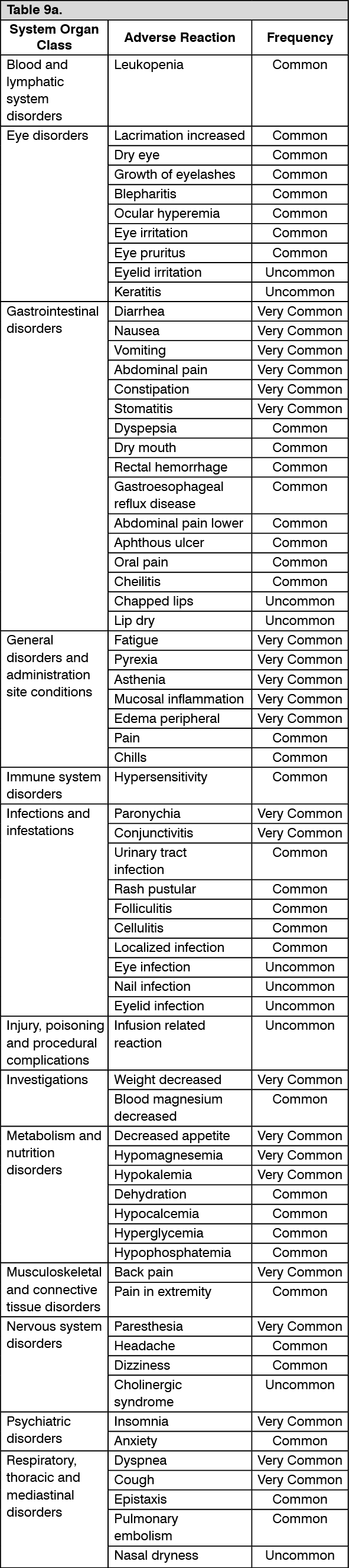 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image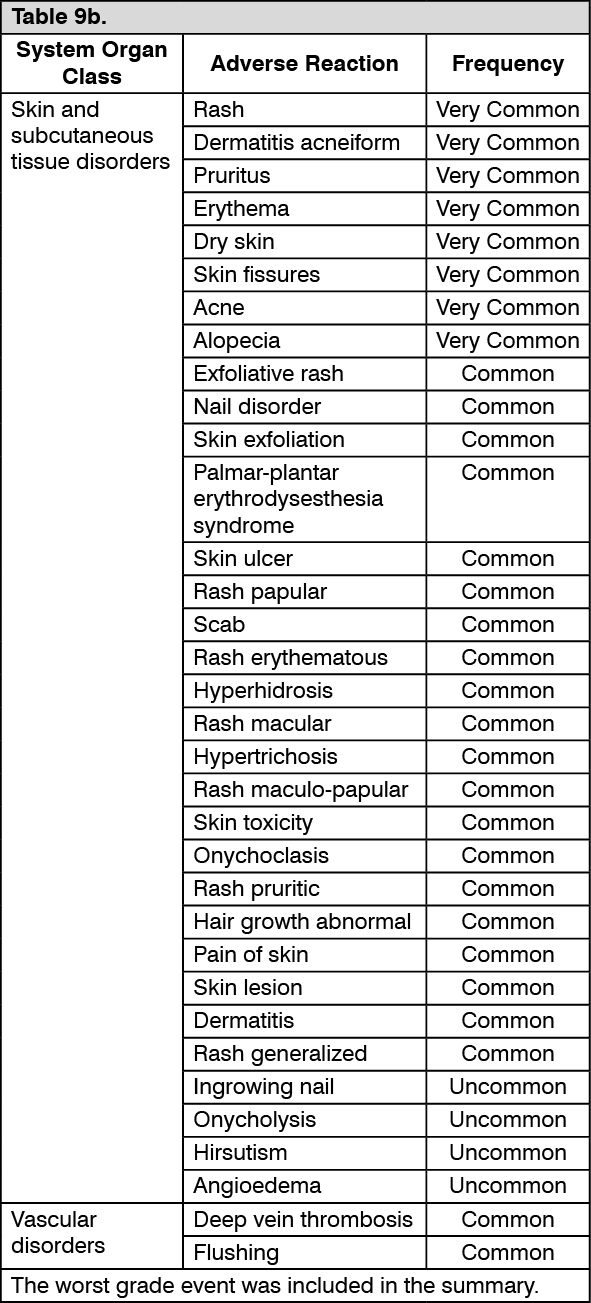 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
