Pharmacotherapeutic group: Anti-neoplastic agents, protein kinase inhibitors.
ATC code: L01XE47.
Pharmacology: Pharmacodynamics: Mechanism of action: Dacomitinib is a pan-human epidermal growth factor receptor (HER) (EGFR/HER1, HER2, and HER4) inhibitor, with activity against mutated EGFR with deletions in exon 19 or the L858R substitution in exon 21. Dacomitinib binds selectively and irreversibly to its HER family targets thereby providing prolonged inhibition.
Clinical efficacy: VIZIMPRO in first-line treatment of NSCLC patients with EGFR-activating mutations (ARCHER 1050): The efficacy and safety of VIZIMPRO was studied in a Phase 3 trial (ARCHER 1050) conducted in patients with locally advanced, not amenable to curative surgery or radiotherapy, or metastatic NSCLC harbouring activating mutations of EGFR, to demonstrate the superiority of dacomitinib versus gefitinib. A total of 452 patients were randomised 1:1 to dacomitinib or gefitinib in a multicentre, multinational, randomised, open-label Phase 3 study.
Treatment was administered orally on a continuous daily basis until disease progression, institution of new anticancer therapy, intolerable toxicity, withdrawal of consent, death, or investigator decision dictated by protocol compliance, whichever occurred first. Stratification factors at randomisation were race (Japanese versus mainland Chinese versus other East Asian versus non-East Asian, as stated by the patient) and EGFR mutation status (exon 19 deletion versus the L858R mutation in exon 21). EGFR mutation status was determined by a standardised and commercially available test kit.
The primary endpoint of the study was progression-free survival (PFS) as determined by blinded Independent Radiology Central (IRC) review. Key secondary endpoints included objective response rate (ORR), duration of response (DoR), and overall survival (OS).
The demographic characteristics of the overall study population were 60% female; median age at enrolment was 62 years with 10.8% being ≥ 75 years old. Thirty percent had baseline Eastern Cooperative Oncology Group (ECOG) performance status (PS) 0 and 70% had ECOG PS 1; 59% had an exon 19 deletion, and 41% had a L858R mutation in exon 21. Race was 23% White, 77% Asian, and < 1% Black. Patients with brain metastases or leptomeningeal disease or ECOG PS ≥ 2 were excluded from the trial.
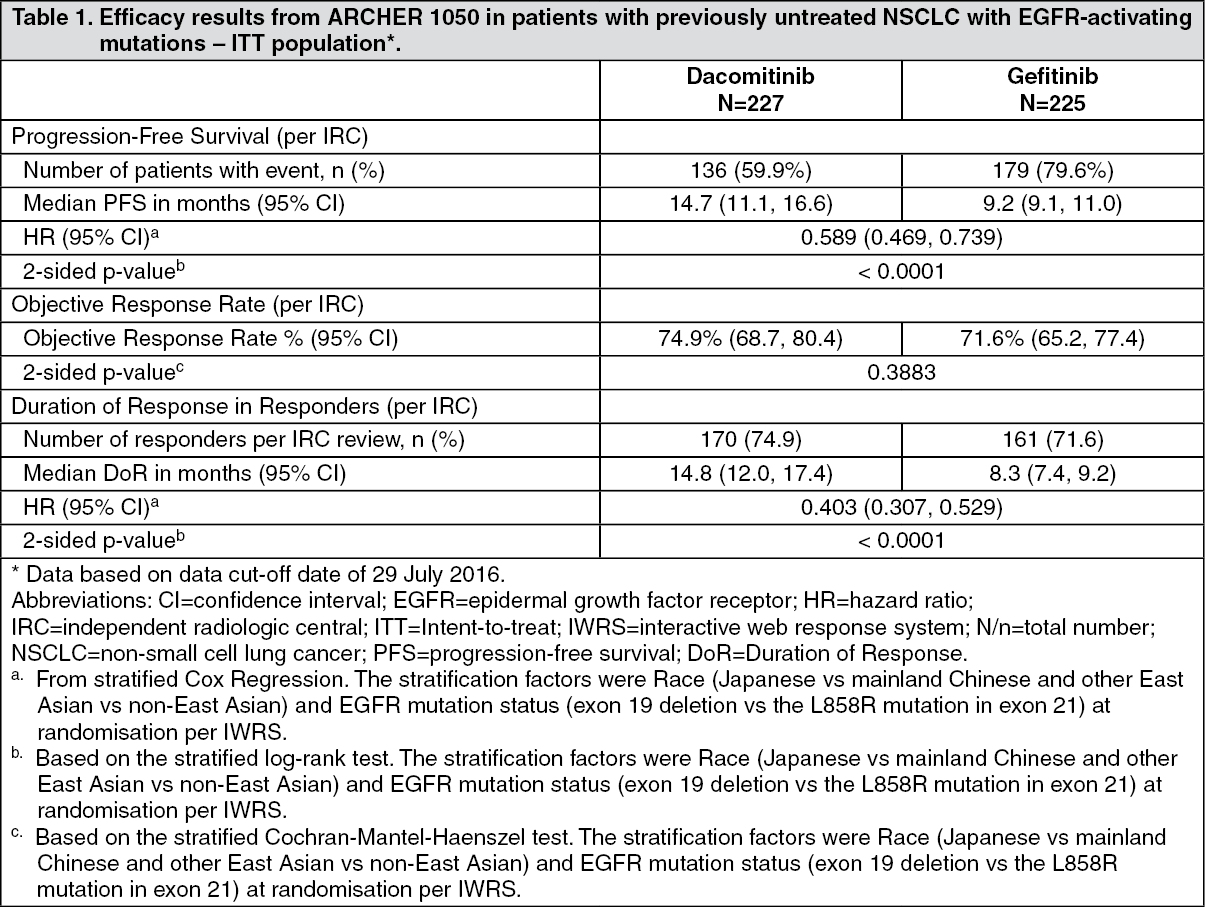
A statistically significant improvement in PFS as determined by the IRC was demonstrated for patients randomised to dacomitinib compared with those randomised to gefitinib, see Table 1 and Figure. Subgroup analyses of PFS per IRC review based on baseline characteristics were consistent with those from the primary analysis of PFS. In particular, the hazard ratios (HRs) for PFS per IRC review in Asian and non-Asian patients were 0.509 (95% CI: 0.391, 0.662) and 0.889 (95% CI: 0.568, 1.391), respectively. In Asian patients, median PFS was 16.5 months for dacomitinib arm and 9.3 months for gefitinib arm. In non-Asian patients, median PFS was 9.3 months for dacomitinib arm and 9.2 months for gefitinib arm.
OS results from the final analysis (data cut-off date of 17-Feb-2017) when 48.7% of events had occurred showed a HR of 0.760 (95% CI: 0.582, 0.993) and a gain of 7.3 months in median OS (median OS: 34.1 months [95% CI: 29.5, 37.7] and 26.8 months [95% CI: 23.7, 32.1] in the dacomitinib and gefitinib arm, respectively). However, according to the hierarchical testing approach, the analysis was stopped with the testing of ORR as the statistical significance was not reached. Therefore, the statistical significance of OS improvement could not be formally assessed. (See Table 1 and Figure.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Pharmacokinetics: Absorption:
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Pharmacokinetics: Absorption: Following the administration of a single 45 mg dose of dacomitinib tablets, the mean oral bioavailability of dacomitinib is 80% (range: 65% to 100%) compared to intravenous administration, with C
max occurring 5 to 6 hours after oral dosing. Following dacomitinib 45 mg daily dosing, steady-state was reached within 14 days. Food does not alter bioavailability to a clinically meaningful extent. Dacomitinib is a substrate for the membrane transport proteins P-gp and BCRP. However, based on the oral bioavailability of 80%, these membrane transport proteins are unlikely to have any impact on dacomitinib absorption.
Distribution: Dacomitinib is extensively distributed throughout the body with a mean steady-state volume of distribution of 27 L/kg (patient of 70 kg) [coefficient of variation (CV%): 18%] following intravenous administration. In plasma, dacomitinib binds to albumin and α
1-acid glycoprotein and the fraction unbound is approximately 2%
in vitro and
ex vivo in healthy volunteers.
Biotransformation: In humans, dacomitinib undergoes oxidation and glutathione conjugation as the major metabolic pathways. Following oral administration of a single 45-mg dose of [
14C] dacomitinib, the most abundant circulating metabolite was O-desmethyl dacomitinib. This metabolite exhibited
in vitro pharmacologic activity that was similar to that of dacomitinib in the
in vitro biochemical assays. In faeces, dacomitinib, O-desmethyl dacomitinib, a cysteine conjugate of dacomitinib, and a mono-oxygenated metabolite of dacomitinib were the major drug-related components.
In vitro studies indicated that CYP2D6 was the major CYP isozyme involved in the formation of O-desmethyl dacomitinib, while CYP3A4 contributed to the formation of other minor oxidative metabolites. O-desmethyl dacomitinib accounted for 16% of human plasma radioactivity and is formed mainly by CYP2D6 and to a lesser extent CYP2C9. The inhibition of CYP2D6 translated into approximately a 90% reduction in metabolite exposure and an approximate 37% increase in dacomitinib exposure.
Other information on drug-drug interactions: Effect of dacomitinib and O-desmethyl dacomitinib on CYP enzymes:
In vitro, dacomitinib and its metabolite O-desmethyl dacomitinib have a low potential to inhibit the activities of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, or CYP3A4/5 at clinically relevant concentrations.
In vitro, dacomitinib has a low potential to induce CYP1A2, CYP2B6, or CYP3A4 at clinically relevant concentrations.
Effect of dacomitinib on drug transporters:
In vitro, dacomitinib has a low potential to inhibit the activities of drug transporters P-gp (systemically), organic anion transporters (OAT)1 and OAT3, OCT2, organic anion transporting polypeptide (OATP)1B1, and OATP1B3, but may inhibit the activity of P-gp (in the GI tract), BCRP (systemically and GI tract), and OCT1 at clinically relevant concentrations.
Effect of Dacomitinib on UGT Enzymes:
In vitro, dacomitinib has a low potential to inhibit uridine-diphosphate glucuronosyltransferase (UGT)1A4, UGT1A6, UGT1A9, UGT2B7, and UGT2B15.
Elimination: The plasma half-life of dacomitinib ranges from 54 to 80 hours. Dacomitinib showed a clearance of 20.0 L/hr with an inter-individual variability of 32% (CV%). In 6 healthy male subjects given a single-oral dose of [
14C] radiolabeled dacomitinib, a median of 82% of the total administered radioactivity was recovered in 552 hours; faeces (79% of dose) was the major route of excretion, with 3% of the dose recovered in urine, of which <1% of the administered dose was unchanged dacomitinib.
Special populations: Age, race, gender, body weight: Based on population pharmacokinetic analyses, patient age, race (Asian and non-Asian), gender and body weight do not have a clinically relevant effect on predicted steady-state exposure of dacomitinib. Approximately 90% of patients included in this analysis were Asian or White.
Hepatic impairment: In a dedicated hepatic impairment trial, following a single-oral dose of 30 mg VIZIMPRO, dacomitinib exposure (AUC
inf and C
max) was unchanged in mild hepatic impairment (Child-Pugh A; N=8) and decreased by 15% and 20%, respectively in moderate hepatic impairment (Child-Pugh B; N=9) when compared to subjects with normal hepatic function (N=8). Dacomitinib pharmacokinetics has not been studied in subjects with severe hepatic impairment (Child-Pugh class C). In addition, based on a population pharmacokinetic analysis using data from 1381 patients, that included 158 patients with mild hepatic impairment defined by National Cancer Institute (NCI) criteria [total bilirubin ≤ Upper Limit of Normal (ULN) and Aspartate Aminotransferase (AST) > ULN, or total bilirubin >1.0 to 1.5 × ULN and any AST; N=158], mild hepatic impairment had no effect on the pharmacokinetics of dacomitinib. From the small number of patients in the moderate group [total bilirubin >1.5 to 3 × ULN and any AST; N=5], there is no evidence for a change in dacomitinib pharmacokinetics.
Renal impairment: No clinical studies have been conducted in patients with impaired renal function. Based on population pharmacokinetic analyses, mild (60 mL/min ≤ CrCl <90 mL/min; N=590) and moderate (30 mL/min ≤ CrCl <60 mL/min; N=218) renal impairment, did not alter dacomitinib pharmacokinetics, relative to subjects with normal (CrCl ≥90 mL/min; N=567) renal function. Limited pharmacokinetic data are available in patients with severe renal impairment (CrCl <30 mL/min) (N=4). The pharmacokinetics in patients requiring haemodialysis have not been studied.
Exposure response relationships: No clear relationship between dacomitinib exposure and efficacy could be characterised over the exposure range studied. Significant exposure-safety relationship was defined for Grade ≥3 rash/dermatitis acneiform, other skin toxicities, diarrhoea and Grade ≥1 stomatitis.
Toxicology: Preclinical safety data: Repeated-dose toxicity: In oral repeated-dose toxicity studies for up to 6 months in rats and 9 months in dogs, the primary toxicities were identified in the skin/hair (dermal changes in rats and dogs, atrophy/dysplasia of hair follicles in rats), kidney (papillary necrosis often accompanied by tubular degeneration, regeneration, dilatation and/or atrophy and changes in urinary markers indicative of renal injury in rats, erosion or ulceration of the pelvic epithelium with associated inflammation without changes indicative of renal dysfunction in dogs), eye (cornea epithelial atrophy in rats and dogs, corneal ulcers/erosions with red/swollen conjunctiva(e), conjunctivitis, elevated third eyelid, increased squinting, partially closed eyes, lacrimation, and/or ocular discharge in dogs), and digestive system (enteropathy in rats and dogs, erosions/ulcers of the mouth with reddened mucous membranes in dogs), and atrophy of epithelial cells of other organs in rats. In addition, hepatocellular necrosis with transaminase increases and hepatocellular vacuolation were observed in rats only. These effects were reversible with the exception of hair follicles and kidney changes. All effects occurred at systemic exposure below that in humans at the recommended dose of 45 mg once daily.
Genotoxicity: Dacomitinib was tested using a series of genetic toxicology assays. Dacomitinib was not mutagenic in a bacterial reverse mutation (Ames) assay, and not clastogenic or aneugenic in the
in vivo bone marrow micronucleus assay in male and female rats. Dacomitinib was clastogenic in the
in vitro human lymphocyte chromosome aberration assay at cytotoxic concentrations.
Dacomitinib is not directly reactive toward DNA as evidenced by the negative response in the bacterial reverse mutation assay and did not induce chromosome damage in a bone marrow micronucleus assay at concentrations up to approximately 60-70 times the unbound AUC or C
max at the recommended human dose. Thus, dacomitinib is not expected to be genotoxic at clinically relevant exposure concentrations.
Carcinogenicity: Carcinogenicity studies have not been performed with dacomitinib.
Impairment of fertility: Fertility studies have not been performed with dacomitinib. In repeat-dose toxicity studies with dacomitinib, effects on reproductive organs were observed in female rats given approximately 0.3 times the unbound AUC at the recommended human dose (for 6 months) and were limited to reversible epithelial atrophy in the cervix and vagina. There was no effect on reproductive organs in male rats given ≤2 mg/kg/day for 6 months (approximately 1.1 times the unbound AUC at the recommended human dose), or in dogs given ≤1 mg/kg/day for 9 months (approximately 0.3 times the unbound AUC at the recommended human dose).
Developmental toxicity: In embryo-foetal development studies in rats and rabbits, pregnant animals received oral doses up to approximately 2.4 times and 0.3 times, respectively, the unbound AUC at the recommended human dose during the period of organogenesis. Maternal body weight gain and food intake were lower in pregnant rats and rabbits. The maternally toxic dose was foetotoxic in rats, resulting in reduced foetal body weights and higher incidence of unossified metatarsals.
Phototoxicity: A phototoxicity study with dacomitinib in pigmented rats showed no phototoxicity potential.
Environmental risk assessment: Environmental risk assessment studies have shown that dacomitinib has the potential to be very persistent, bioaccumulative and toxic to the environment (see Cautions for Usage).


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image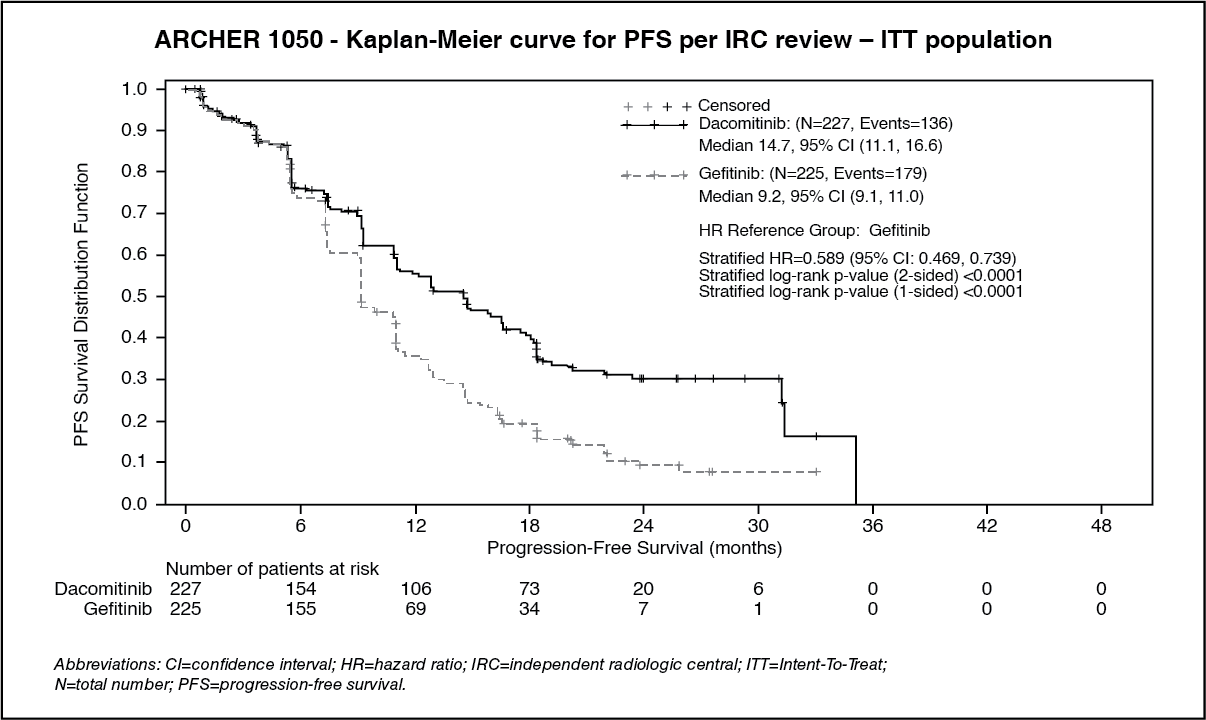 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image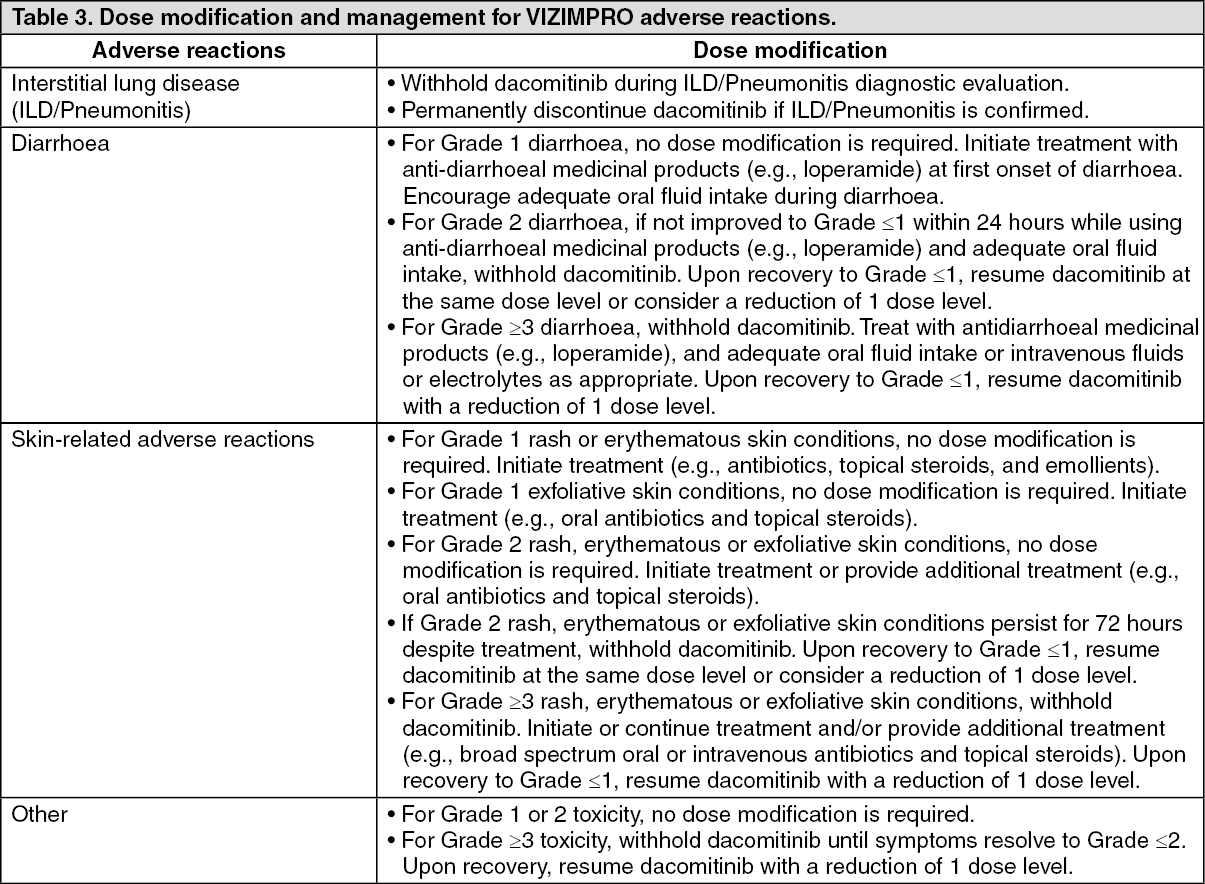 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image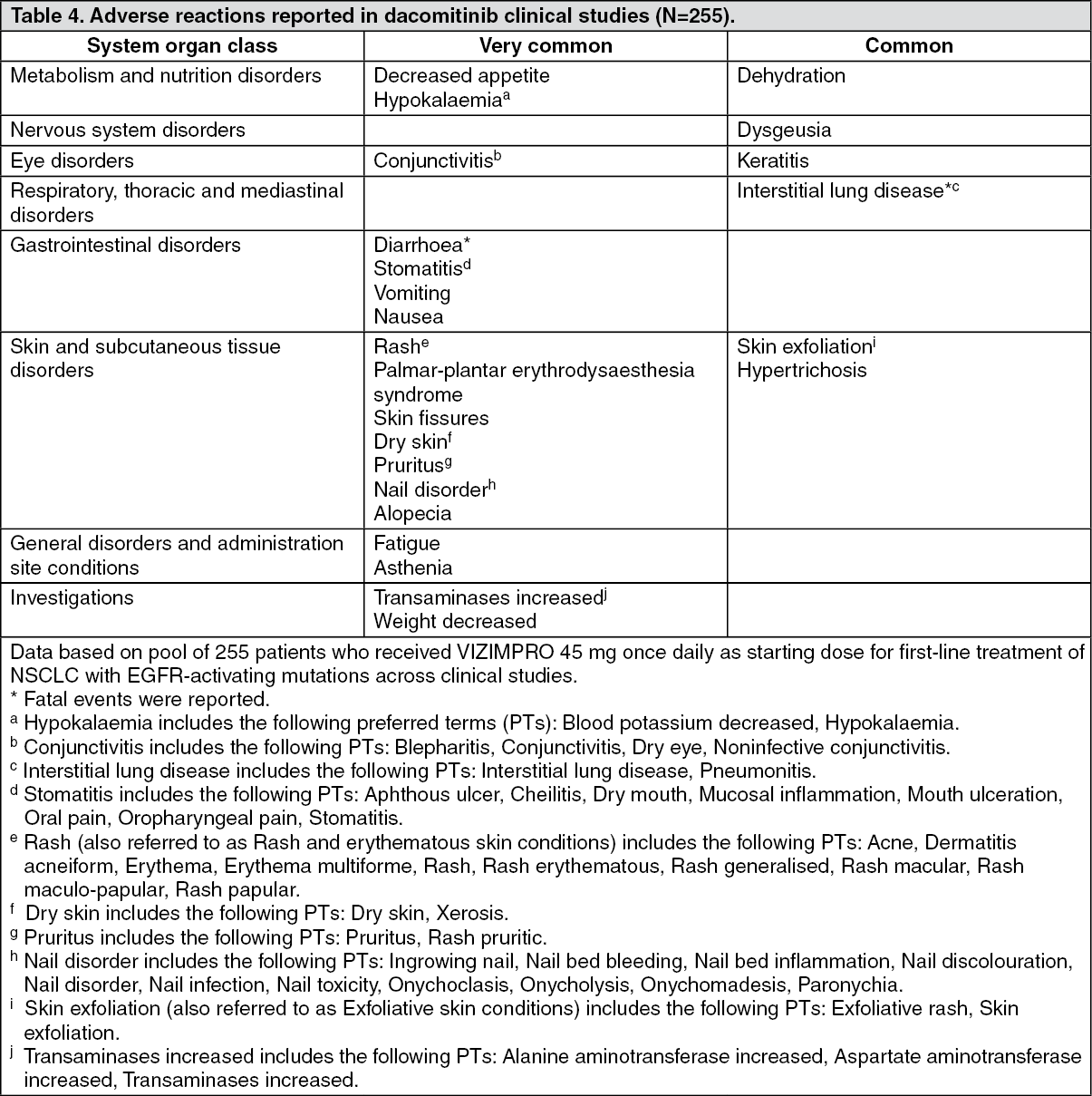 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image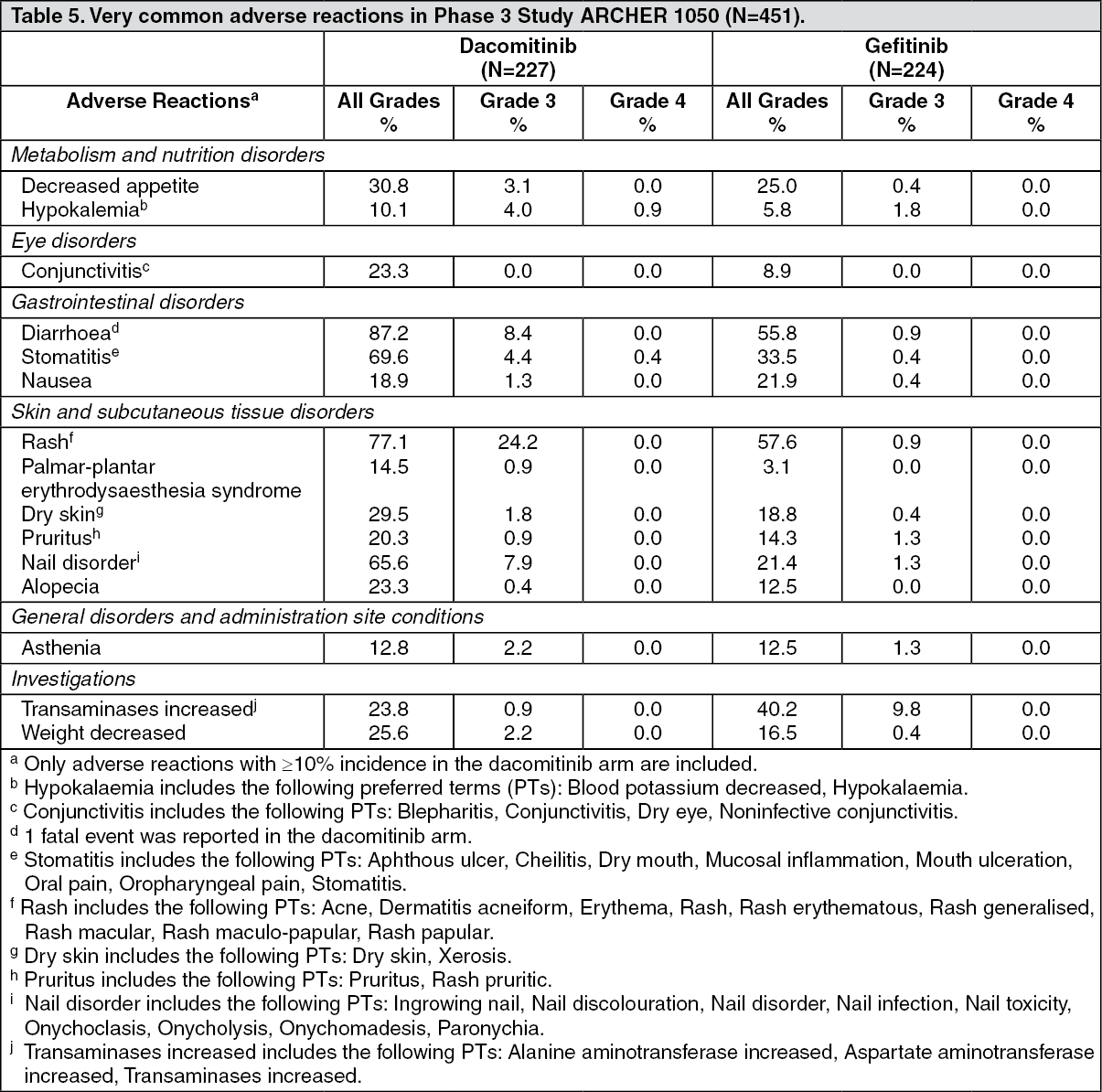 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
