Pharmacotherapeutic Group: Antineoplastic agents - Protein kinase inhibitor.
ATC Code: L01XE11.
Pharmacology: Pharmacodynamics: Mechanism of Action (MOA): Pazopanib is an orally administered, potent multi-target tyrosine kinase inhibitor (TKI) of vascular endothelial growth factor receptors (VEGFR)-1, -2, and -3, platelet-derived growth factor (PDGFR)-alpha and -beta, and stem cell factor receptor (c-KIT), with IC
50 values of 10, 30, 47, 71, 84 and 74 nM, respectively. In preclinical experiments, pazopanib dose-dependently inhibited ligand-induced auto-phosphorylation of VEGFR-2, c-Kit and PDGFR-beta receptors in cells. In vivo, pazopanib inhibited VEGF-induced VEGFR-2 phosphorylation in mouse lungs, angiogenesis in various animal models, and the growth of multiple human tumor xenografts in mice.
Pharmacogenomics: In a pharmacogenetic meta-analysis of data from 31 clinical studies of Votrient administered either as monotherapy or in combination with other agents, ALT >5 X ULN (NCI CTC Grade 3) occurred in 19% of HLA-B*57:01 allele carriers and in 10% of non-carriers. In this dataset, 133/2235 (6%) of the patients carried the HLA-B*57:01 allele (see Precautions).
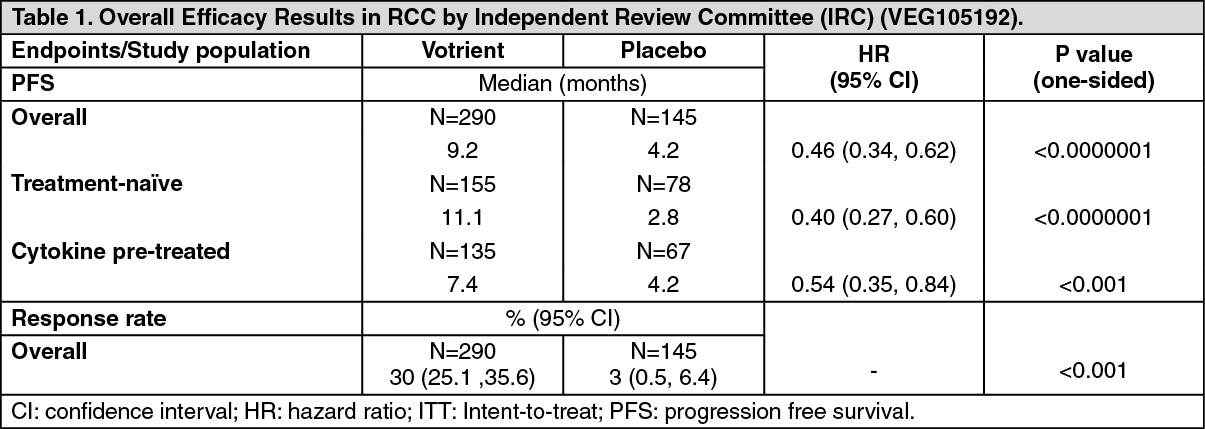
Clinical Studies: Renal Cell Carcinoma (RCC): The safety and efficacy of Votrient in renal cell carcinoma (RCC) were evaluated in a randomized, double-blind, placebo-controlled multi-center study. Patients (N=435) with locally advanced and/or metastatic RCC were randomized to receive Votrient 800 mg once daily or placebo. The primary objective of the study was to evaluate and compare the two treatment arms for progression-free survival (PFS) and the principle secondary endpoint is overall survival (OS). The other objectives were to evaluate the overall response rate and duration of response.
From the total of 435 patients in this study, 233 patients were treatment naïve and 202 were second line patients who had received one prior IL-2 or INF-alpha-based therapy. The performance status (ECOG) was similar between the Votrient and placebo groups (ECOG 0: 42% vs. 41%, ECOG 1: 58% vs. 59%). All patients had clear cell histology or predominantly clear cell histology. Approximately half of all patients had 3 or more organs involved in their disease and most patients had the lung (74%), and/or lymph nodes (54%) as a metastatic location for disease at baseline.
A similar proportion of patients in each arm were treatment-naïve and cytokine-pre-treated (53% and 47% in Votrient arm, 54% and 46% in placebo arm). In the cytokine-pre-treated subgroup, the majority (75%) had received interferon based treatment.
Similar proportions of patients in each arm had prior nephrectomy (89% and 88% in the Votrient and placebo arms, respectively) and/or prior radiotherapy (22% and 15% in the Votrient and placebo arms, respectively).
The primary analysis of the primary endpoint PFS is based on disease assessment by independent radiological review in the entire study population (first line and second line). (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
For patients who responded to treatment, the median duration of response was 58.7 weeks as per independent review. The median overall survival (OS) data at the protocol specified final survival analysis were 22.9 months and 20.5 months [HR=0.91 (95% CI: 0.71, 1.16; p=0.224)] for patients randomized to the Votrient and placebo arms, respectively. The OS results are subject to potential bias as 54% of patients in the placebo arm also received Votrient in the extension part of this study following disease progression. Sixty-six percent of placebo patients received post-study therapy compared to 30% of Votrient patients.
In the pivotal study, the QoL assessments were based on blinded self-reported global scores from two protocol-specified questionnaires, EORTC QLQ-C30 and EuroQoL EQ-5D. Analysis was based on patients who continued on therapy in both arms, prior to progression. The assessments showed no difference between treatment with Votrient or placebo (p >0.05), indicating no negative impact of Votrient on global quality of life.
In a Phase II study of 225 patients with locally recurrent or metastatic clear cell renal cell carcinoma, objective response rate was 35% and median duration of response was 68 weeks, as per independent review.
The safety, efficacy and quality of life of Votrient versus sunitinib was evaluated in a randomized, open-label, parallel group Phase III non-inferiority study (VEG108844).
In VEG108844, patients (N=1,110) with locally advanced and/or metastatic RCC who had not received prior systemic therapy, were randomized to receive either Votrient 800 mg once daily continuously or sunitinib 50 mg once daily in 6-week cycles of dosing with 4 weeks on treatment followed by 2 weeks without treatment.
The primary objective of this study was to evaluate and compare PFS in patients treated with Votrient to those treated with sunitinib. Demographic characteristics were similar between the treatment arms. Disease characteristics at initial diagnosis and at screening were balanced between the treatment arms with the majority of patients having clear cell histology and Stage IV disease.
VEG108844 achieved its primary endpoint of PFS and demonstrated that Votrient was non-inferior to sunitinib, as the upper bound of the 95% CI for the hazard ratio was less than the protocol-specified non-inferiority margin of 1.25. Overall efficacy results are summarized in Table 2. (See Table 2.)
Click on icon to see table/diagram/image
Soft tissue sarcoma (STS): The safety and efficacy of Votrient in STS were evaluated in a randomized, double-blind, placebo-controlled multi-center study. Patients (N=369) with advanced STS who had received prior chemotherapy, including anthracycline treatment, or were unsuited for such therapy, were randomized to receive Votrient 800 mg once daily or placebo.
Prior to randomization, eligible subjects were stratified by the factors of WHO performance status (WHO PS) (0 or 1) at baseline and the number of lines of prior systemic therapy for advanced disease (0 or 1 vs. 2+). In each treatment group, there were a slightly greater percentage of subjects in the 2+ lines of prior systemic therapy for advanced disease (58% and 55% respectively for placebo and Votrient treatment arms) compared with 0 or 1 lines of prior systemic therapy (42% and 45% respectively for placebo and Votrient treatment arms). There were slightly more subjects with a WHO PS of 1 at baseline. The median duration of follow-up of subjects (defined as date of randomization to date of last contact or death) was similar for both treatment arms (9.36 months for placebo [range 0.69 to 23.0 months] and 10.04 months for Votrient [range 0.2 to 24.3 months].
The primary objective of the study was to evaluate and compare the two treatment arms for progression-free survival (PFS), based on the ITT population, and the principle secondary endpoint is overall survival (OS).
The initial analysis of the primary endpoint PFS was based on disease assessment by independent radiological review in the entire ITT study population. (See Table 3.)
Click on icon to see table/diagram/image
Similar to the assessments by independent radiology review, a clinically meaningful and statistically significant improvement in PFS based on investigator assessments was observed in the Votrient arm compared with the placebo arm (HR: 0.39; 95% CI, 0.30 to 0.52, p <0.001).
The hazard ratio at the pre-specified interim analysis for overall survival in favor of Votrient was not statistically significant; the median overall survival in the placebo arm was 10.4 months (95% CI 8.7 to 12.7) and was 11.9 months (95% CI 10.7 to 15.1) in the Votrient arm; HR=0.82 (97.87% CI: 0.59 to 1.14, p=0.156).
Pharmacokinetics: Absorption: Pazopanib is absorbed orally with median time to achieve peak concentrations of 2.0 to 4.0 hours after the dose. Daily dosing results in 1.23- to 4-fold increase in AUC. There was no consistent increase in AUC and C
max when the Votrient dose increased above 800 mg once daily.
Systemic exposure to pazopanib is increased when administered with food. Administration of Votrient with a high-fat or low-fat meal results in an approximately 2-fold increase in AUC and C
max. Therefore, Votrient should be administered at least 1 hour before or 2 hours after a meal (see Dosage & Administration and Interactions).
Administration of a single Votrient 400 mg crushed tablet increased AUC
(0-72) by 46% and C
max by approximately 2-fold and decreased tmax by approximately 1.5 hours compared to administration of the whole tablet. These results indicate that the bioavailability and the rate of pazopanib oral absorption are increased after administration of the crushed tablet relative to administration of the whole tablet. Therefore, due to this potential for increased exposure, tablets should not be crushed (see Dosage & Administration).
Distribution: Binding of pazopanib to human plasma protein in vivo was greater than 99% with no concentration dependence over the range of 10 to 100 microgram/mL. In vitro studies suggest that pazopanib is a substrate for P-glycoprotein (P-gp) and breast cancer resistant protein (BCRP).
Biotransformation/metabolism: Results from in vitro studies demonstrated that the metabolism of Votrient is mediated primarily by CYP3A4, with minor contributions from CYP1A2 and CYP2C8.
Elimination: Pazopanib is eliminated slowly with mean half-life of 30.9 hours after administration of the recommended dose of 800 mg. Elimination is primarily via feces with renal elimination accounting for <4% of the administered dose.
Special Populations: Renal Impairment: In a population pharmacokinetic analysis using 408 subjects with various cancers, creatinine clearance (30 to 150 mL/min) did not influence clearance of pazopanib. Renal impairment is not expected to influence pazopanib exposure, and dose adjustment is not necessary in patients with creatinine clearance ≥30 mL/min (see Dosage & Administration).
Hepatic Impairment: The median steady-state pazopanib C
max and AUC
(0-24) in patients with mild hepatic impairment (defined as either normal bilirubin and any degree of ALT elevations or as an elevation of bilirubin up to 1.5 X ULN regardless of the ALT value) after a once daily dose of 800 mg/day (30.9 microgram/mL, range 12.5 to 47.3 and 841.8 microgram.hr/mL, range 600.4 to 1,078) are similar to the median in patients with no hepatic impairment (49.4 microgram/mL, range 17.1 to 85.7 and 888.2 microgram.hr/mL, range 345.5 to 1,482) (see Dosage & Administration).
The maximally tolerated Votrient dose (MTD) in patients with moderate hepatic impairment (defined as an elevation of bilirubin >1.5 X to 3 X ULN regardless of the ALT values) was 200 mg once daily. The median steady-state values of C
max (22.4 microgram/mL, range 6.4 to 32.9) and AUC
(0-24) (350.0 microgram.hr/mL, range 131.8 to 487.7) after administration of 200 mg Votrient once daily in subjects with moderate hepatic impairment were approximately 45% and 39%, respectively, that of the corresponding median values after administration of 800 mg once daily in subjects with normal hepatic function (see Dosage & Administration).
There are insufficient data in patients with severe hepatic impairment (total bilirubin >3 X ULN regardless of the ALT value); therefore, use of Votrient is not recommended in these patients.
Toxicology: Non-Clinical Safety Data: Carcinogenicity and mutagenicity: In two year carcinogenicity studies with pazopanib, there were increased numbers of liver adenomas noted in mice and duodenal adenocarcinomas noted in rats. Based on the rodent-specific pathogenesis and mechanism for these findings, they are not considered to represent an increased carcinogenic risk for patients taking Votrient.
Pazopanib did not cause genetic damage when tested in genotoxicity assays (Ames assay, human peripheral lymphocyte chromosome aberration assay, and rat
in vivo micronucleus assay).
Fertility: In female rats, reduced fertility (including increased pre- and post-implantation loss and early resorptions) was noted at dosages ≥10 mg/kg/day (approximately 0.2-fold the AUC at the MRHD of 800 mg/day). Decreased corpora lutea were noted in monkeys given 500 mg/kg/day for up to 34 weeks, in mice given ≥100 mg/kg/day for 13 weeks and ovarian atrophy was noted in rats given 300 mg/kg/day for 26 weeks (approximately equal to, 0.6, 1.4 and 0.9-fold the AUC at the MRHD of 800 mg/day, respectively).
Pazopanib did not affect mating or fertility in male rats. However, there were reductions in sperm production rates, sperm motility, and epididymal and testicular sperm concentrations observed at ≥100 mg/kg/day (approximately 0.5-fold the AUC at the MRHD of 800 mg/day) following 15 weeks of dosing. Following 26 weeks of dosing, there were decreased testicular and epididymal weights, atrophy and degeneration of the testes with aspermia, hypospermia and cribiform change in the epididymis of male rats given doses ≥30 mg/kg/day (approximately 0.4-fold the AUC at the MRHD of 800 mg/day).
Safety pharmacology and repeat dose toxicity: In toxicology studies in rats, there were effects in a variety of tissues (bone, teeth, bone marrow, nail beds, reproductive organs, hematological tissues, kidney, adrenal glands, lymph node, pituitary, and pancreas) consistent with VEGFR inhibition and/or disruption of VEGF signalling pathways with some effects occurring at doses of 3 mg/kg/day (approximately 0.1-fold the AUC at the MRHD of 800 mg/day).
Hepatic effects included mild elevations of liver transaminases in rodents and bilirubin elevations in monkeys without associated histopathology at doses that produced systemic exposures approximately 0.1 and 0.6 times the human clinical exposure, respectively.
Reproductive toxicity: For information on reproductive toxicity, see Use in Pregnancy & Lactation.
Juvenile animal studies: In juvenile toxicity studies, when pre-weaning rats were dosed from day 9 postpartum through day 14 postpartum, pazopanib caused mortalities and abnormal organ growth/maturation in kidney, lung, liver and heart, at a dose approximately 0.1-fold the AUC at the MRHD of 800 mg/day. When post weaning rats were dosed from day 21 postpartum to day 62 postpartum, toxicological findings were similar to adult rats at comparable exposures with changes in bone, trachea, teeth, adrenal, pancreas, stomach, duodenum, lymph node, male mammary gland and reproductive organs. In rats, weaning occurs at day 21 postpartum which approximately equates to a human pediatric age of 2 years. Human pediatric patients are at increased risk for bone and teeth effects as compared to adults, as these changes, including shortened limbs, were present in juvenile rats at ≥10 mg/kg/day (equal to approximately 0.1- 0.2-fold the AUC at the MRHD of 800 mg/day) (see Precautions).


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image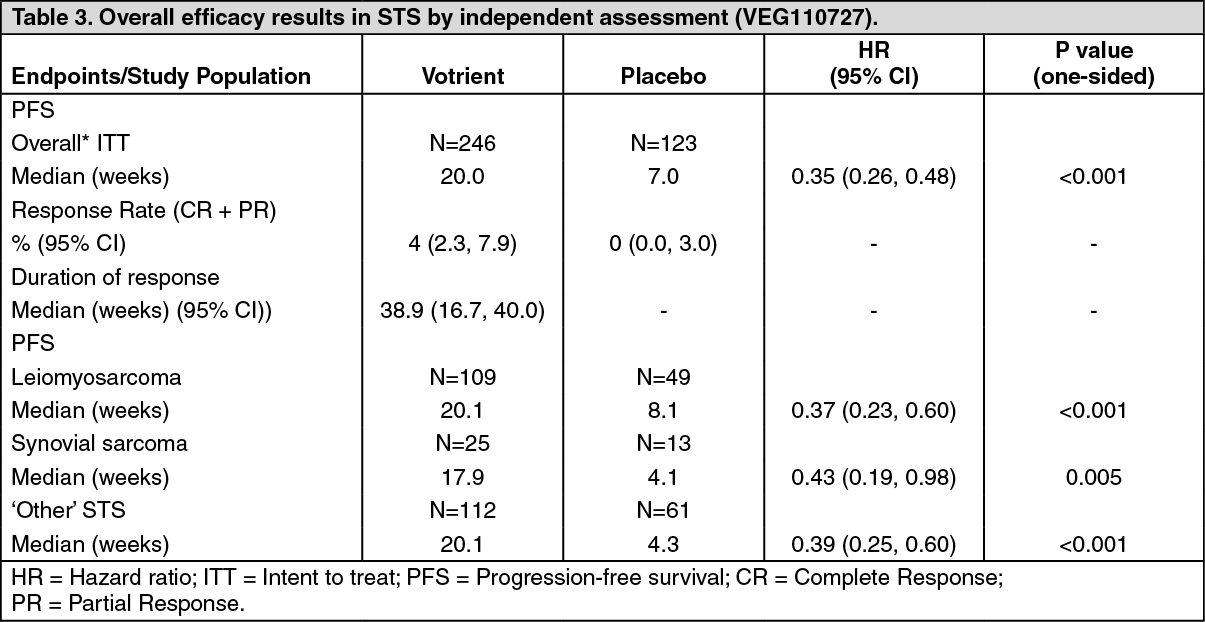 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image


 Sign Out
Sign Out
