PHARMACOLOGY: Pharmacodynamics: Mechanism of action: Xospata is a small molecule FMS-like tyrosine kinase 3 (FLT3) and AXL tyrosine kinase inhibitor. Xospata demonstrated the ability to inhibit FLT3 receptor signaling and proliferation in cells exogenously expressing FLT3 including FLT3-ITD, FLT3-D835Y, and FLT3-ITD-D835Y, and it induced apoptosis in leukemic cells expressing FLT3-ITD.
Pharmacodynamic effects: In patients with relapsed or refractory AML receiving Xospata 120 mg, substantial (> 90%) inhibition of FLT3 phosphorylation was rapid (within 24 hours after first dose) and sustained, as characterised by an
ex vivo plasma inhibitory activity (PIA) assay.
Prolonged QT interval: A concentration-related increase in change from baseline of QTcF (ΔQTcF) was observed across Xospata doses ranging from 20 to 450 mg. The predicted mean change from baseline of QTcF at the mean steady-state C
max (282.0 ng/mL) at the 120 mg daily dose was 4.96 msec with an upper 1-sided 95% CI = 6.20 msec. Of 317 patients treated with Xospata at 120 mg with a post-baseline QTc value in clinical trials, 4 patients (<1.3%) experienced a QTcF >500 msec.
Additionally, across all doses, 2.3% of patients with relapse/refractory AML had a maximum post-baseline QTcF interval >500 msec.
Clinical efficacy and safety: ADMIRAL study (2215-CL-0301): The ADMIRAL trial is a Phase 3, open-label, multicenter, randomized clinical trial of adult patients with relapsed or refractory AML and a FLT3 mutation. FLT3 mutations were identified by a diagnostic test. The trial compares the safety and efficacy of gilteritinib therapy (120 daily dose) to one of the following salvage chemotherapies: cytarabine 20 mg twice daily by subcutaneous (SC) or intravenous (IV) for 10 days (days 1 through 10); azacitidine 75 mg/m
2 once daily by SC or IV for 7 days (days 1 through 7); mitoxantrone 8 mg/m
2, etoposide 100 mg/m
2 and cytarabine 1000 mg/m
2 once daily by IV for 5 days (days 1 through 5); granulocyte colony-stimulating factor 300 mcg/m
2 once daily by SC for 5 days (days 1 to 5), fludarabine 30 mg/m
2 once daily by IV for 5 days (days 2 through 6), cytarabine 2000 mg/m
2 once daily by IV for 5 days (days 2 through 6), idarubicin 10 mg/m
2 once daily by IV for 3 days (days 2 through 4).
Patients included were relapsed or refractory after first line AML therapy and were stratified by response to prior AML treatment and preselected chemotherapy i.e. high or low intensity. While the study included patients with various AML-related cytogenetic abnormalities, patients with acute promyelocytic leukaemia (APL) or therapy-related AML were excluded.
Sixteen patients were randomised but not treated in the study (1 patient in the gilteritinib arm and 15 patients in the chemotherapy arm). Gilteritinib was given orally at a starting dose of 120 mg daily until unacceptable toxicity or lack of clinical benefit. Dose reductions were allowed, to manage adverse reactions, and dose increases were allowed, for those patients who did not respond at the starting dose of 120 mg.
Of the patients who were pre-selected to receive salvage chemotherapy, 60.5% were randomised to high intensity and 39.5% to low intensity. MEC and FLAG-Ida were given for up to two cycles depending on response to first cycle. LoDAC and azacitidine were given in continuous 4-week cycles until unacceptable toxicity or lack of clinical benefit.
The efficacy of gilteritinib was evaluated in a pre-planned interim analysis of 142 patients that were randomized to the gilteritinib arm. At the final analysis, overall survival (OS) was evaluated in 371 patients randomized in a 2:1 ratio (247 in the gilteritinib arm and 124 in the salvage chemotherapy arm).
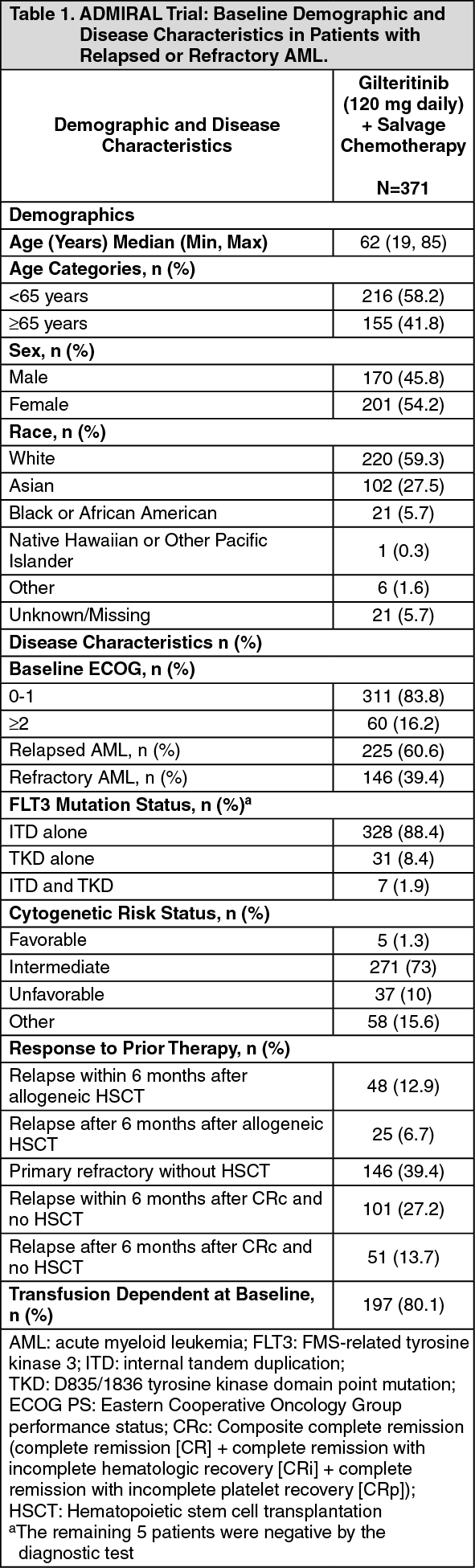
The baseline demographic and disease characteristics are shown in the table as follows. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The primary efficacy endpoint for the final analysis was OS, measured from the date of randomization until death by any cause (number of events analyzed was 261). Patients randomized to the gilteritinib arm had significantly longer survival compared to the chemotherapy arm (HR 0.637; 95% CI 0.490 - 0.830; 1 sided p-value: 0.0004). The median OS was 9.3 months for patients receiving gilteritinib and 5.6 months for those receiving chemotherapy (Table 2, figure). (See Table 2 and figure.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
A modified analysis of event free survival (EFS), defined as a failure to obtain a composite complete remission (CRc) with failures assigned as an event on date of randomization, relapse, or death from any cause, including events and initiation of new anti-leukemia treatments reported in long-term follow up, showed an improvement with a median EFS of 2.3 months for gilteritinib versus 0.7 months for salvage chemotherapy HR 0.499 (95% CI 0.387, 0.643) and 1-sided p<0.0001.
Efficacy was supported by the rate of complete remission (CR)/complete remission with partial hematologic recovery (CRh), the duration of CR/CRh (DOR), and the rate of conversion from transfusion dependence to transfusion independence. The efficacy results are shown in the table as follows. (See Table 3.)
Click on icon to see table/diagram/image
For patients who achieved a CR/CRh, the median time to first response was 3.7 months (range: 0.9 to 10.6 months) in the gilteritinib arm and 1.2 months (range: 1 to 2.6 months) in the salvage chemotherapy arm. The median time to best response of CR/CRh was 3.8 months (range: 0.9 to 16 months) in the gilteritinib arm and 1.2 months (range: 1 to 2.6 months) in the salvage chemotherapy arm.
Among the 197 patients who were dependent on red blood cell (RBC) and/or platelet transfusions at baseline, 68 (34.5%) became independent of RBC and platelet transfusions during any 56-day post-baseline period. For the 49 patients who were independent of both RBC and platelet transfusions at baseline, 29 (59.2%) remained transfusion independent during any 56-day post-baseline period.
CHRYSALIS Trial (2215-CL-0101): The efficacy of gilteritinib was evaluated in an open-label, multicenter, dose escalation, clinical trial (CHRYSALIS trial) investigating the safety, tolerability, pharmacokinetics and pharmacodynamics of gilteritinib therapy in patients with relapsed or refractory AML and a FLT3 mutation. FLT3 mutations were identified by local results.
The baseline demographic and disease characteristics are shown in the table as follows. (See Table 4.)
Click on icon to see table/diagram/image
Efficacy was established on the basis of the rate of CR/CRh, the duration of CR/CRh (DOR), and the rate of conversion from transfusion dependence to transfusion independence. Efficacy for patients who received gilteritinib at the 120 mg daily dose level are represented in the table as follows. (See Table 5.)
Click on icon to see table/diagram/image
For patients who achieved a CR/CRh, the median time to first response of CR/CRh was 1.9 months (range: 1 to 9.2 months), and the median time to best response of CR/CRh was 2.1 months (range: 1 to 12 months).
Among the 52 patients who were dependent on red blood cell (RBC) and/or platelet transfusions at baseline, 11 (21.2%) became independent of RBC and platelet transfusions during any 56-day post-baseline period. Of the 4 patients who were independent of both RBC and platelet transfusions at baseline, 3 (75.0%) remained transfusion independent during any 56-day post-baseline period.
Pharmacokinetics: Absorption: Following oral administration of tablet formulations, peak Xospata concentrations are observed at a median t
max approximately between 4 and 6 hours in healthy volunteers and patients with relapsed or refractory AML. Xospata undergoes first-order absorption with an estimated absorption rate (k
a) of 0.43 hr
-1 with a lag time of 0.34 hours based on population pharmacokinetic (PK) modeling. Median steady-state maximum concentration (C
max) is 282.0 ng/mL (CV% = 50.8), and area under the plasma concentration curve during 24-hour dosing interval (AUC
0-24) is 6180 ng·hr/mL (CV% = 46.4) after once-daily dosing of 120 mg Xospata. Steady-state plasma levels are reached by 15 days of once-daily dosing with an approximate 10-fold accumulation.
Effect of food: In healthy adults, Xospata C
max and AUC decreased by approximately 26% and less than 10%, respectively, when a single 40 mg dose of Xospata was coadministered with a high fat meal compared to Xospata exposure in fasted state. Median t
max was delayed 2 hours when Xospata was administered with a high-fat meal. Xospata can be administered with or without food.
Distribution: The population estimates of central and peripheral volume of distribution were 1092 L and 1100 L, respectively. These data indicate Xospata distributes extensively outside of plasma, which may indicate extensive tissue distribution.
In vivo plasma protein binding in humans is approximately 90% and Xospata is primarily bound to albumin.
Biotransformation: Based on
in vitro data, Xospata is primarily metabolised via CYP3A4. The primary metabolites in humans include M17 (formed via N-dealkylation and oxidation), M16 and M10 (both formed via N-dealkylation) and were observed in animals. None of these three metabolites exceeded 10% of overall parent exposure.
Elimination: After a single dose of [
14C]-Xospata, Xospata is primarily excreted in feces with 64.5% of the total administered dose recovered in feces. Renal excretion is a minor elimination pathway with 16.4% of the total dose recovered in urine as unchanged drug and metabolites. Xospata plasma concentrations declined in a bi-exponential manner with a population mean estimated half-life of 113 hours. The estimated apparent clearance (CL/F) based on the population PK model is 14.85 L/h.
Linearity: Xospata exhibits linear, dose-proportional pharmacokinetics in patients with relapsed or refractory AML at doses ranging from 20 mg to 450 mg administered once-daily.
Special populations: No clinically meaningful effect on the pharmacokinetics of Xospata was observed for the following covariates: age (20 years to 90 years), race (Caucasian, Black, Asian or Other), mild hepatic impairment [defined as total bilirubin ≤ upper limit of normal (ULN) and aspartate transaminase (AST) >ULN or total bilirubin 1 to 1.5 times ULN and any AST], sex, body weight (36 kg to 157 kg), and body surface area (1.29 to 2.96 m
2).
Hepatic impairment: The effect of hepatic impairment on Xospata pharmacokinetics has been studied in subjects with mild (Child-Pugh Class A) and moderate (Child-Pugh Class B) hepatic impairment. Results indicate unbound Xospata exposure in subjects with mild or moderate hepatic impairment is comparable to that observed in subjects with normal hepatic function. Xospata has not been studied in patients with severe hepatic impairment (Child-Pugh Class C).
In non-AML patients with mild (Child-Pugh Class A) or moderate (Child-Pugh Class B) hepatic impairment, unbound Xospata exposure is comparable to that observed in subjects with normal liver function. The effect of mild hepatic impairment on Xospata exposure was also assessed using the population PK model and the results demonstrate little difference in predicted steady-state Xospata exposure relative to a typical patient with relapsed or refractory AML normal liver function. These data suggest Xospata dose adjustment is not warranted in patients with mild or moderate hepatic impairment. Xospata has not been studied in patients with severe hepatic impairment (Child-Pugh Class C).
Renal impairment: A clinical assessment of the effect of renal function on Xospata exposure was not conducted based on nonclinical and clinical data that indicate renal excretion is a minor elimination route. Although the population PK model included serum creatinine, a marker of renal function, as a statistically significant covariate, the impact on Xospata exposure was less than 2-fold and not considered clinically meaningful. Therefore, impaired renal function is not expected to significantly affect Xospata exposure indicating dose adjustment is not warranted in patients with renal impairment.
Drug Interaction Studies: Effects of Other Drugs on gilteritinib: CYP3A/P-gp Inducers: Gilteritinib exposure decreased approximately 70% when gilteritinib was coadministered with a strong CYP3A/P-gp inducer (rifampicin). Concomitant medications that are strong CYP3A/P-gp inducers should be avoided during gilteritinib therapy.
CYP3A and/or P-gp Inhibitors: Gilteritinib exposure increased approximately 2.2-fold when gilteritinib was coadministered with a strong CYP3A and P-gp inhibitor (itraconazole) in healthy adult subjects and approximately 1.5-fold in patients with relapsed or refractory AML. It is recommended that concomitant medications that are strong CYP3A and/or P-gp inhibitors be used with caution during gilteritinib therapy as they can increase the plasma exposure of gilteritinib.
In vitro experiments demonstrated that gilteritinib is a substrate of BCRP.
Effects of gilteritinib on Other Drugs: Based on
in vitro data, gilteritinib may reduce the effects of drugs that target 5HT
2B receptor or sigma nonspecific receptor. Avoid concomitant use of these drugs with gilteritinib unless use is considered essential for the care of the patient.
Gilteritinib may potentially inhibit BCRP, P-gp and OCT1 at clinically relevant concentrations.
Toxicology: Preclinical safety data: Gilteritinib showed a concentration-dependent suppression effect on the human ether-a-go-go related gene (hERG) current in hERG-transfected HEK293 cells. The IC50 was 1.6 x 10-5 mol/L (8.84 mcg/mL).
Gilteritinib increased the currents via CaV1.2 calcium channel in Chinese hamster ovary cells and KV7.1/minK potassium channel in HEK293 cells at 1 x 10-6 mol/L (553 ng/mL) and higher concentrations.
In rats treated with gilteritinib, decreased urination at 30 mg/kg and higher and decreased defecation at 100 mg/kg were observed. In dogs treated with gilteritinib, retching at 3 mg/kg, vomiting and positive fecal occult blood at 10 mg/kg and higher, a decrease in the blood calcium concentration at 30 mg/kg, and salivation and an increase followed by a decrease in the blood calcium concentration at 100 mg/kg were observed.
No repeat-dose studies with dosing duration longer than 13 weeks and no carcinogenicity studies have been conducted.
Gilteritinib did not induce gene mutation in the
in vitro reversion test in bacteria. Similarly, gilteritinib did not induce chromosomal aberrations in the
in vitro chromosomal aberration test in mammalian cells. The
in vivo micronucleus test showed that gilteritinib has a potential to induce micronuclei in mice.
Gilteritinib showed no potential to induce phototoxicity to cultured mammalian cells.
Gilteritinib showed suppressed fetal growth, embryo-fetal deaths and teratogenicity in the embryo-fetal development studies in rats. The no-observed adverse effect level (NOAEL) for dams and embryo-fetal development was 10 mg/kg per day. No embryo-fetal development study in rabbits was conducted.
The effects on pre- and post-natal development and maternal function are unknown.
In the pivotal juvenile animal toxicity study in rats, dosing from postnatal day 4 to 42, the minimum lethal dose level was 2.5 mg/kg per day which was lower than the 20 mg/kg per day dose, the minimal lethal dose in adult rats. In the preliminary (non-GLP) dose range finding study (dosing from postnatal day 4 to up to day 21), gastrointestinal bleeding suggested by abnormal stool color (dark red) was noted at 10 mg/kg per day and higher, indicating that the gastrointestinal tract is one of the target organs as in adult rats.
Single oral administration of [
14C] gilteritinib to pregnant rats resulted in transfer of radioactivity to the fetus similar to that observed in maternal plasma on day 14 of gestation. In addition, distribution profiles of radioactivity in most maternal tissues and the fetus on day 18 of gestation were similar to that on day 14 of gestation.
After single oral administration to lactating rats, milk concentrations of radioactivity were higher than radioactivity in maternal plasma at 4- and 24-hours post-dose and no radioactivity was detected in all tested maternal samples at 48 hours or later post-dose. The radioactivity was detected in the infant tissues examined, except for the brain, at 4, 24, 48, and 72 h post-dose, indicating that gilteritinib-derived components are distributed to the infant tissues through breast milk.
In the 1-week oral repeated dose toxicity study in rats, interstitial pneumonia in the lung and vacuolar change in the rod-cone layer of the retina were observed at 30 mg/kg per day. In the 13-week oral repeated dose toxicity study in rats, deaths occurred at 20 mg/kg per day. Target organ toxicity was identified in the gastrointestinal tract, lymphohematopoietic system, eye, lung, kidney and liver. The no-observed adverse effect level (NOAEL) was lower than 2.5 mg/kg per day.
In the 4-week oral repeated dose study in dogs, mortality/moribundity occurred at 10 mg/kg per day or more. Target organ toxicity was identified in the gastrointestinal tract, lymphohematopoietic system, eye, kidney and liver. The NOAEL was 1 mg/kg per day.
In the 13-week oral repeated dose study in dogs, mortality occurred at 5 mg/kg per day. Target organ toxicity was identified in the lung, urinary bladder, epithelial tissue, gastrointestinal tract, lymphohematopoietic system, eye, kidney and liver. The NOAEL was 1 mg/kg per day.
Reversibility of most of the test article-related changes was indicated by the end of the 4-week recovery period.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image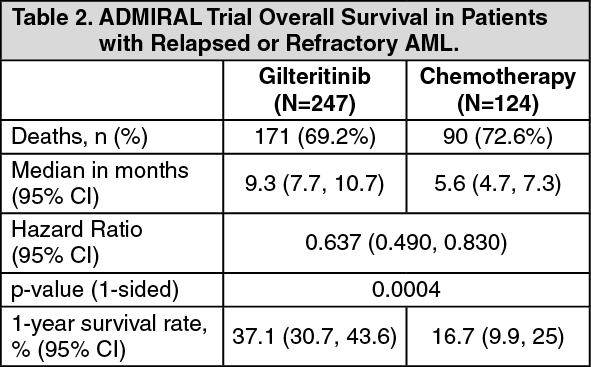 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image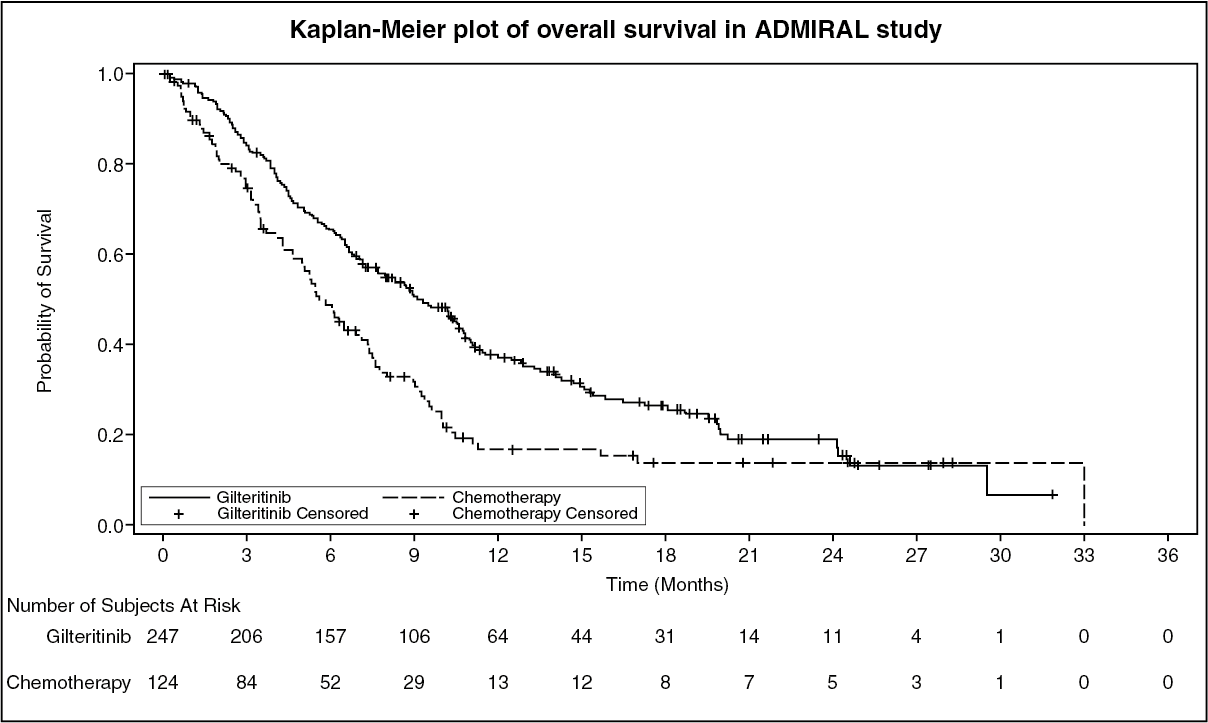 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image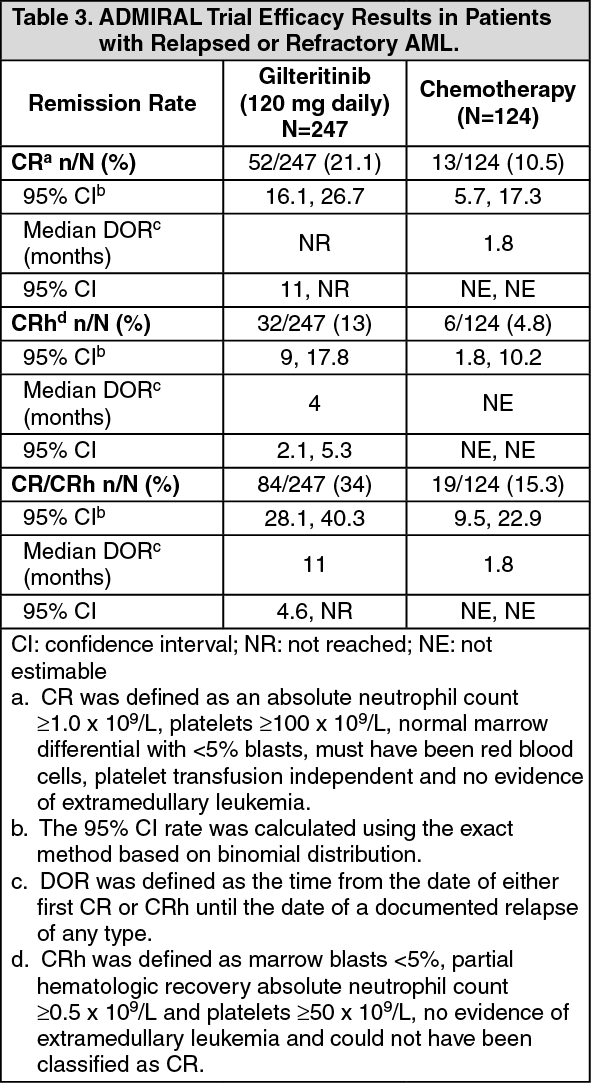 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image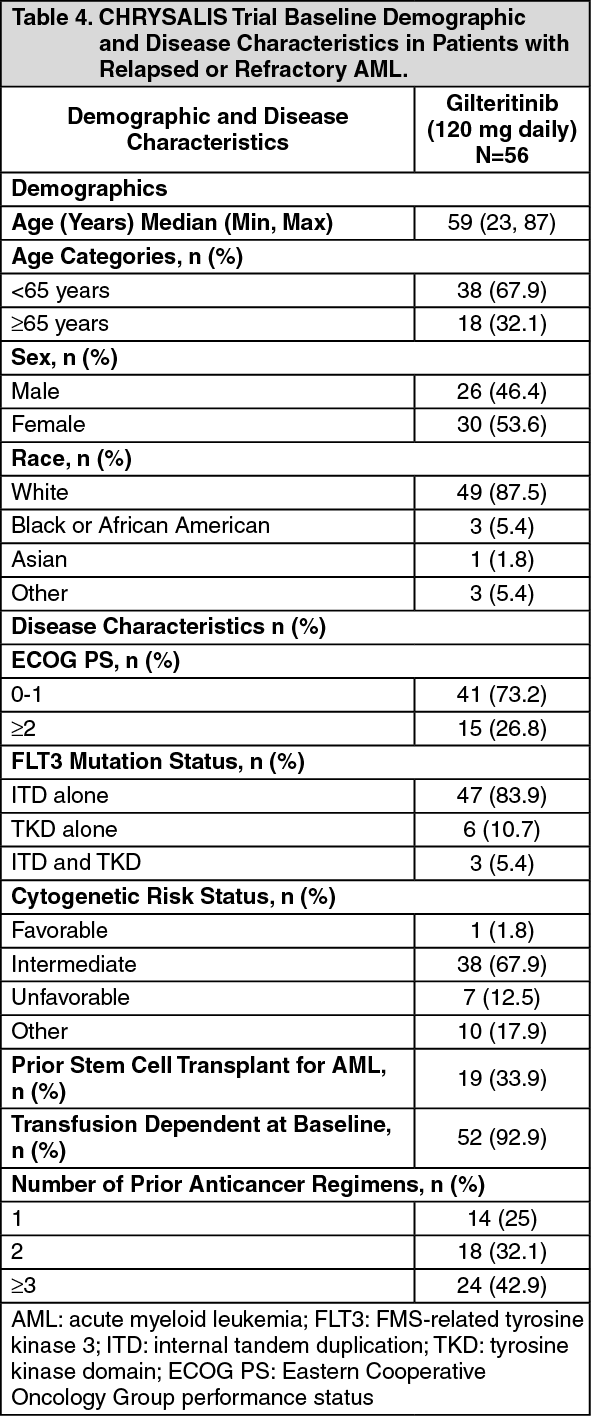 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image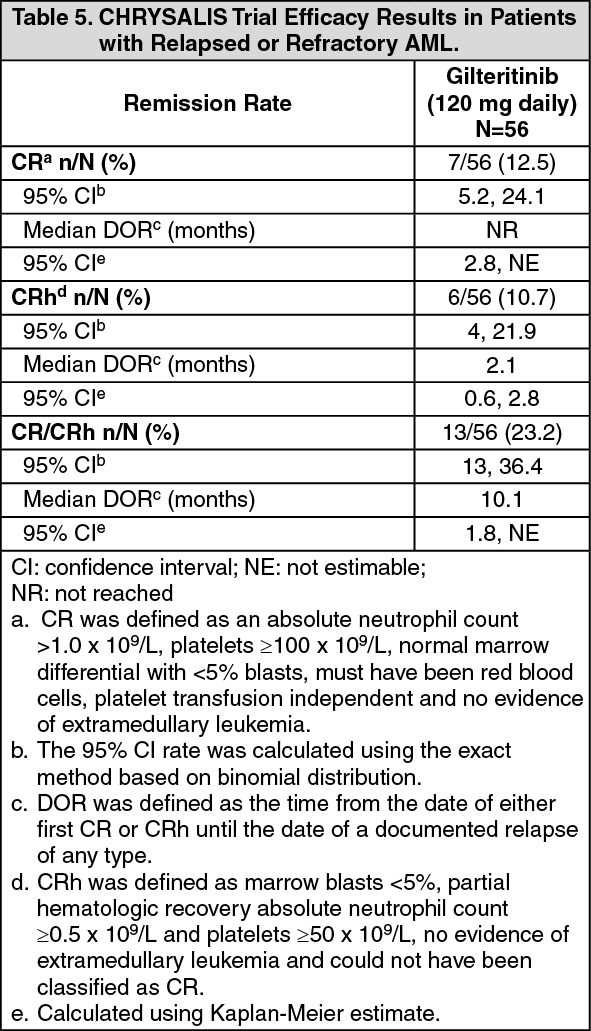 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
