


 Sign Out
Sign Out


Pharmacology: Pharmacodynamics: Solution for SC injection: DARZALEX solution for subcutaneous injection contains recombinant human hyaluronidase (rHuPH20). rHuPH20 works locally and transiently to degrade hyaluronan ((HA), a naturally occurring glycoaminoglycan found throughout the body) in the extracellular matrix of the subcutaneous space by cleaving the linkage between the two sugars (N-acetylglucosamine and glucuronic acid) which comprise HA. rHuPH20 has a half-life in skin of less than 30 minutes. Hyaluronan levels in subcutaneous tissue return to normal within 24 to 48 hours because of the rapid biosynthesis of hyaluronan.
Mechanism of action: Concentrate for solution for IV infusion: Daratumumab is an IgG1κ human monoclonal antibody (mAb) that binds to the CD38 protein expressed at a high level on the surface of multiple myeloma tumour cells, as well as other cell types and tissues at various levels. CD38 protein has multiple functions such as receptor mediated adhesion, signalling and enzymatic activity.
Solution for SC injection: Daratumumab is an IgG1κ human monoclonal antibody (mAb) that binds to the CD38 protein expressed on the surface of cells in a variety of haematological malignancies, including clonal plasma cells in multiple myeloma and AL amyloidosis, as well as other cell types and tissues. CD38 protein has multiple functions such as receptor mediated adhesion, signalling and enzymatic activity.
Concentrate for solution for IV infusion & Solution for SC injection: Daratumumab has been shown to potently inhibit the in vivo growth of CD38-expressing tumour cells. Based on in vitro studies, daratumumab may utilise multiple effector functions, resulting in immune mediated tumour cell death. These studies suggest that daratumumab can induce tumour cell lysis through complement-dependent cytotoxicity, antibody-dependent cell-mediated cytotoxicity, and antibody-dependent cellular phagocytosis in malignancies expressing CD38. A subset of myeloid derived suppressor cells (CD38+MDSCs), regulatory T cells (CD38+Tregs) and B cells (CD38+Bregs) are decreased by daratumumab mediated cell lysis. T cells (CD3+, CD4+, and CD8+) are also known to express CD38 depending on the stage of development and the level of activation. Significant increases in CD4+ and CD8+ T cell absolute counts, and percentages of lymphocytes, were observed with daratumumab treatment in peripheral whole blood and bone marrow. In addition, T-cell receptor DNA sequencing verified that T-cell clonality was increased with daratumumab treatment, indicating immune modulatory effects that may contribute to clinical response.
Daratumumab induced apoptosis in vitro after Fc mediated cross-linking. In addition, daratumumab modulated CD38 enzymatic activity, inhibiting the cyclase enzyme activity and stimulating the hydrolase activity. The significance of these in vitro effects in a clinical setting, and the implications on tumour growth, are not well-understood.
Pharmacodynamic effects: Natural killer (NK) cell and T-cell count: NK cells are known to express high levels of CD38 and are susceptible to daratumumab mediated cell lysis. Decreases in absolute counts and percentages of total NK cells (CD16+CD56+) and activated (CD16+CD56dim) NK cells in peripheral whole blood and bone marrow were observed with daratumumab treatment. However, baseline levels of NK cells did not show an association with clinical response.
Immunogenicity: Concentrate for solution for IV infusion: In patients treated with intravenous daratumumab in clinical trials, less than 1% of patients developed treatment-emergent anti-daratumumab antibodies.
Solution for SC injection: In multiple myeloma and AL amyloidosis patients treated with subcutaneous daratumumab in monotherapy and combination clinical studies, less than 1% of patients developed treatment-emergent anti-daratumumab antibodies.
In multiple myeloma and AL amyloidosis patients, the incidence of treatment-emergent non-neutralizing anti-rHuPH20 antibodies was 7.3% (55/750) in patients who received either monotherapy DARZALEX subcutaneous formulation or combination DARZALEX subcutaneous formulation. The anti-rHuPH20 antibodies did not appear to impact daratumumab exposures. The clinical relevance of the development of anti-daratumumab or anti-rHuPH20 antibodies after treatment with DARZALEX subcutaneous formulation is not known.
Clinical experience with daratumumab concentrate for solution for infusion (intravenous formulation): Newly diagnosed multiple myeloma: Combination treatment with lenalidomide and dexamethasone in patients ineligible for autologous stem cell transplant: Study MMY3008, an open-label, randomised, active-controlled Phase III study, compared treatment with DARZALEX 16 mg/kg in combination with lenalidomide and low-dose dexamethasone (DRd) to treatment with lenalidomide and low-dose dexamethasone (Rd) in patients with newly diagnosed multiple myeloma. Lenalidomide (25 mg once daily orally on Days 1-21 of repeated 28-day [4-week] cycles) was given with low dose oral or intravenous dexamethasone 40 mg/week (or a reduced dose of 20 mg/week for patients >75 years or body mass index [BMI] <18.5). On DARZALEX infusion days, the dexamethasone dose was given as a pre-infusion medicinal product. Dose adjustments for lenalidomide and dexamethasone were applied according to manufacturer's prescribing information. Treatment was continued in both arms until disease progression or unacceptable toxicity.
A total of 737 patients were randomised: 368 to the DRd arm and 369 to the Rd arm. The baseline demographic and disease characteristics were similar between the two treatment groups. The median age was 73 (range: 45-90) years, with 44% of the patients ≥75 years of age. The majority were white (92%), male (52%), 34% had an Eastern Cooperative Oncology Group (ECOG) performance score of 0, 49.5% had an ECOG performance score of 1 and 17% had an ECOG performance score of ≥2. Twenty-seven percent had International Staging System (ISS) Stage I, 43% had ISS Stage II and 29% had ISS Stage III disease. Efficacy was evaluated by progression free survival (PFS) based on International Myeloma Working Group (IMWG) criteria.
Study MMY3008 showed an improvement in Progression Free Survival (PFS) in the DRd arm as compared to the Rd arm; the median PFS had not been reached in the DRd arm and was 31.9 months in the Rd arm (hazard ratio [HR]=0.56; 95% CI: 0.43, 0.73; p<0.0001), representing 44% reduction in the risk of disease progression or death in patients treated with DRd. Results of an updated PFS analysis approximately 9 months after the original clinical cutoff, continued to show an improvement in PFS for patients in the DRd arm compared with the Rd arm. Median PFS was not reached in the DRd arm and was 33.8 months in the Rd arm (HR=0.56; 95% CI: 0.44, 0.71; p<0.0001). (See Figure 1.)
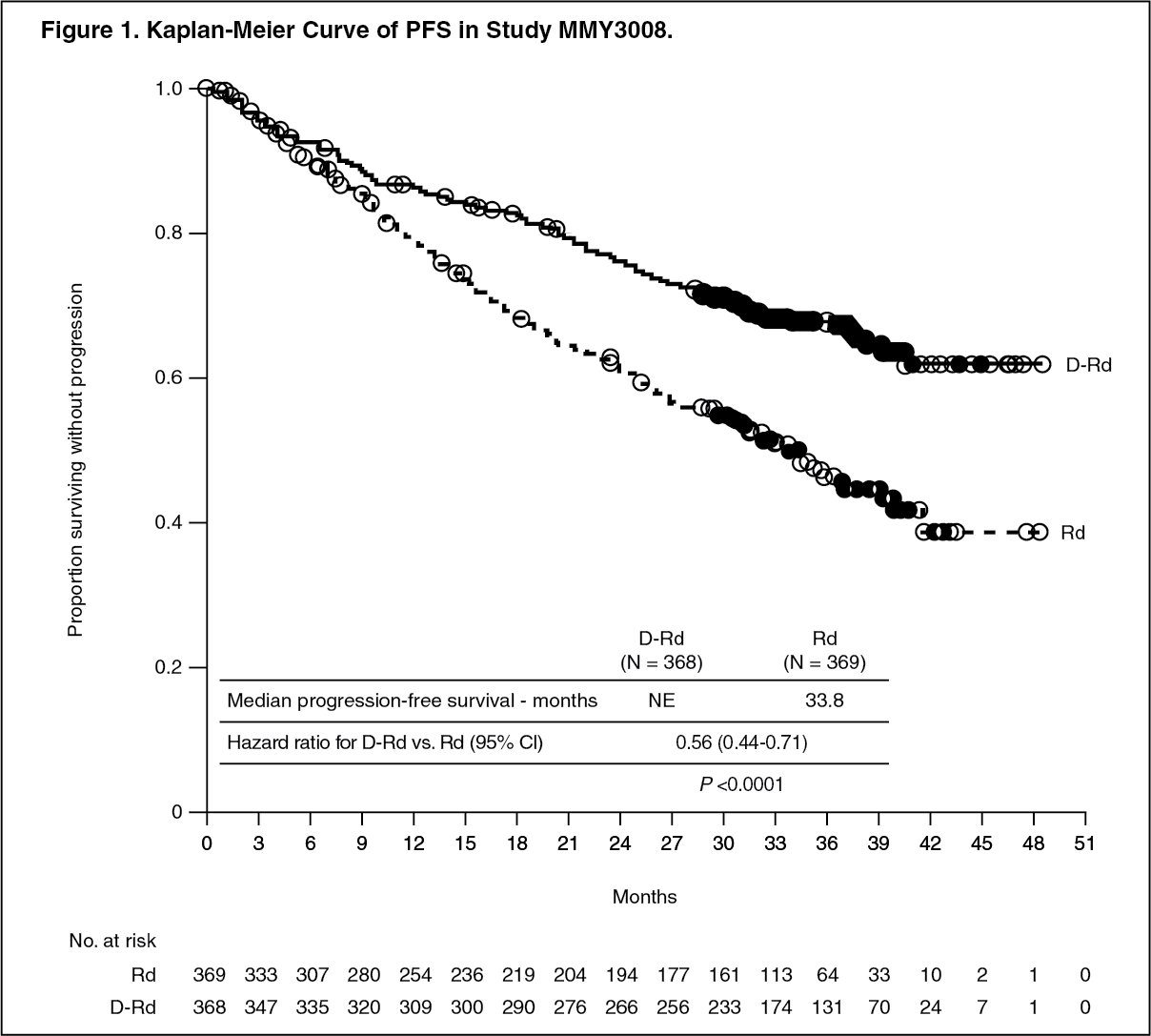 Click on icon to see table/diagram/image
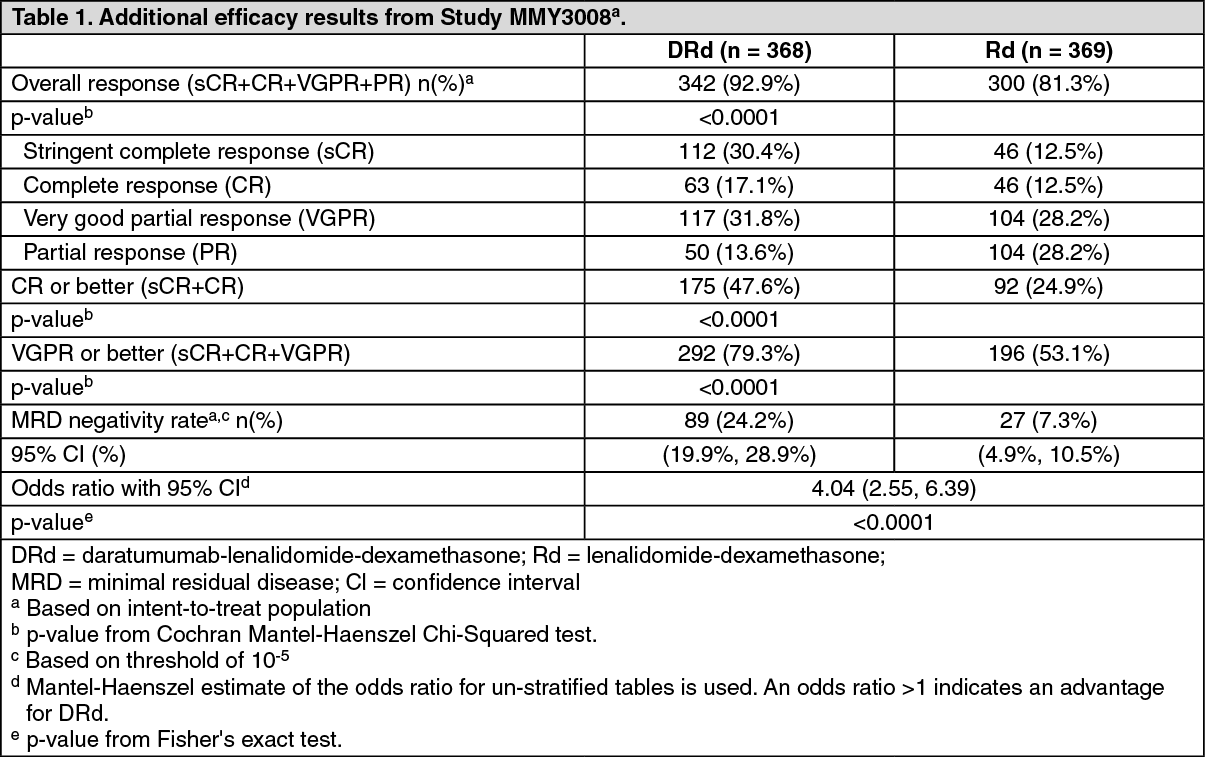
Click on icon to see table/diagram/imageAdditional efficacy results from Study MMY3008 are presented in Table 1 as follows. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn responders, the median time to response was 1.0 months (range: 0.2 to 12.1 months) in the DRd group and 1.05 months (range: 0.3 to 15.3 months) in the Rd group. The median duration of response had not been reached in the DRd group and was 34.7 months (95% CI: 30.8, not estimable) in the Rd group.
Combination treatment with bortezomib, melphalan and prednisone (VMP) in patients ineligible for autologous stem cell transplant: Study MMY3007, an open-label, randomised, active-controlled Phase III study, compared treatment with intravenous DARZALEX 16 mg/kg in combination with bortezomib, melphalan and prednisone (D-VMP), to treatment with VMP in patients with newly diagnosed multiple myeloma. Bortezomib was administered by subcutaneous injection at a dose of 1.3 mg/m2 body surface area twice weekly at Weeks 1, 2, 4 and 5 for the first 6-week cycle (Cycle 1; 8 doses), followed by once weekly administrations at Weeks 1, 2, 4 and 5 for eight more 6-week cycles (Cycles 2-9; 4 doses per cycle). Melphalan at 9 mg/m2, and prednisone at 60 mg/m2 were orally administered on Days 1 to 4 of the nine 6-week cycles (Cycles 1-9). Intravenous DARZALEX treatment was continued until disease progression or unacceptable toxicity.
A total of 706 patients were randomised: 350 to the D-VMP arm and 356 to the VMP arm. The baseline demographic and disease characteristics were similar between the two treatment groups. The median age was 71 (range: 40-93) years, with 30% of the patients ≥75 years of age. The majority were white (85%), female (54%), 25% had an ECOG performance score of 0, 50% had an ECOG performance score of 1 and 25% had an ECOG performance score of 2. Patients had IgG/IgA/Light chain myeloma in 64%/22%/10% of instances, 19% had ISS Stage I, 42% had ISS Stage II, 38% had ISS Stage III disease and 84% had standard risk cytogenetics. Efficacy was evaluated by PFS based on IMWG criteria and overall survival (OS).
With a median follow‑up of 16.5 months, the primary analysis of PFS in Study MMY3007 showed an improvement in the D-VMP arm as compared to the VMP arm; the median PFS had not been reached in the D-VMP arm and was 18.1 months in the VMP arm (HR=0.5; 95% CI: 0.38, 0.65; p<0.0001). Results of an updated PFS analysis after a median follow-up of 40 months continued to show an improvement in PFS for patients in the D-VMP arm compared with the VMP arm. Median PFS was 36.4 months in the D‑VMP arm and 19.3 months in the VMP arm (HR=0.42; 95% CI: 0.34, 0.51; p<0.0001), representing a 58% reduction in the risk of disease progression or death in patients treated with D‑VMP. (See Figure 2.)
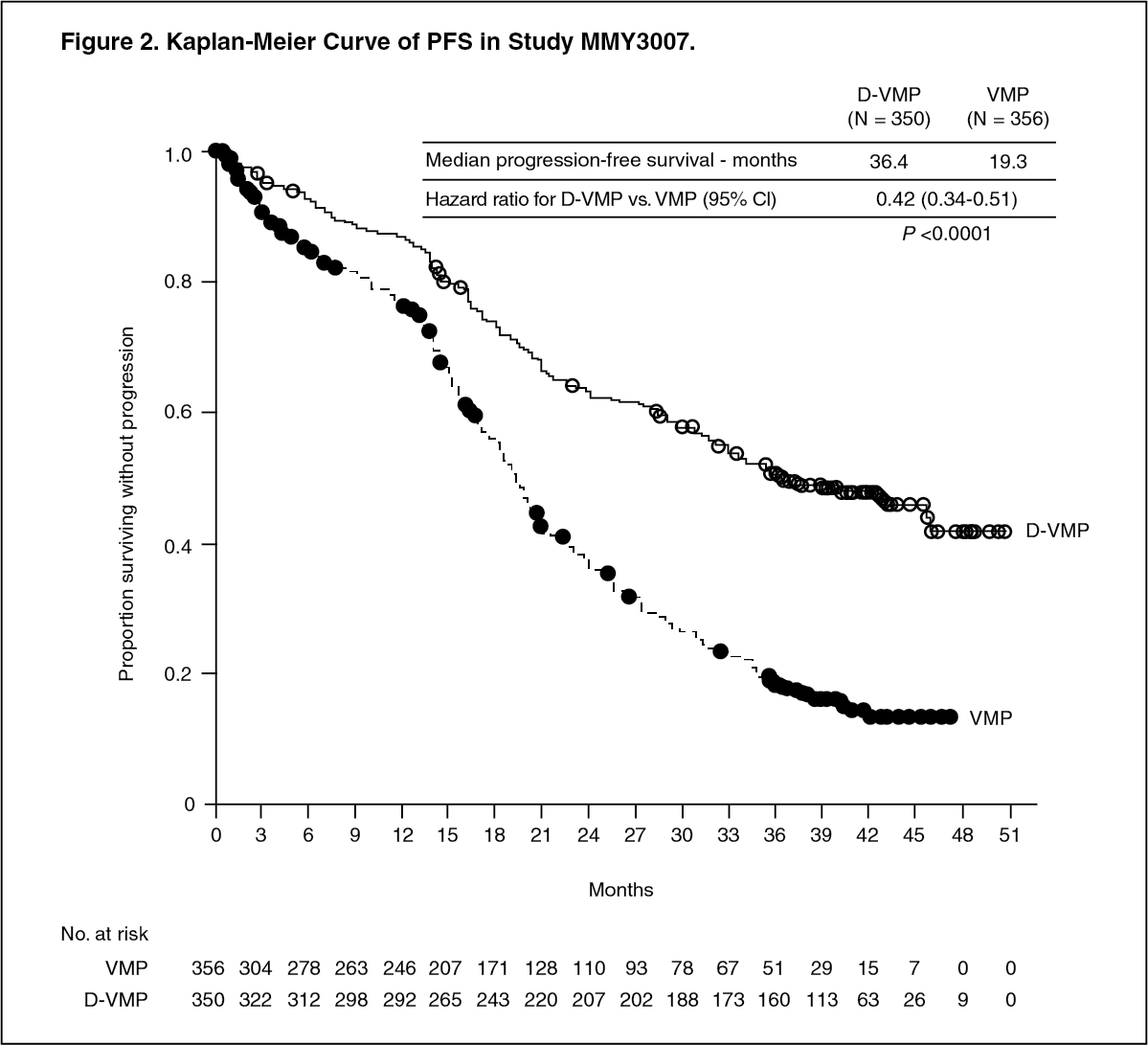 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAfter a median follow‑up of 40 months, D‑VMP has shown an overall survival (OS) advantage over the VMP arm (HR=0.60; 95% CI: 0.46, 0.80; p=0.0003), representing a 40% reduction in the risk of death in patients treated in the D‑VMP arm. Median OS was not reached for either arm. (See Figure 3.)
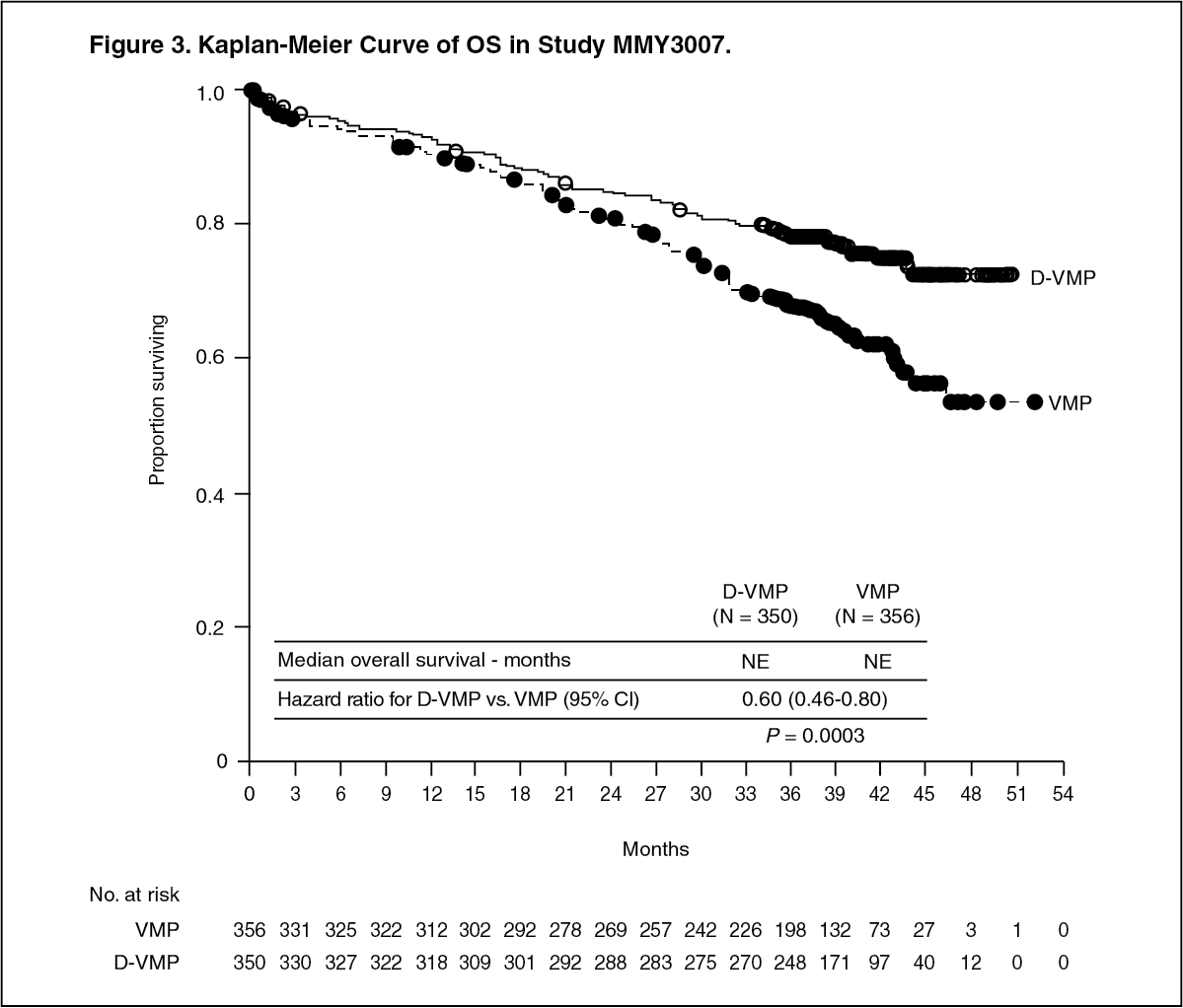 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAdditional efficacy results from Study MMY3007 are presented in Table 2 as follows. (See Table 2.)
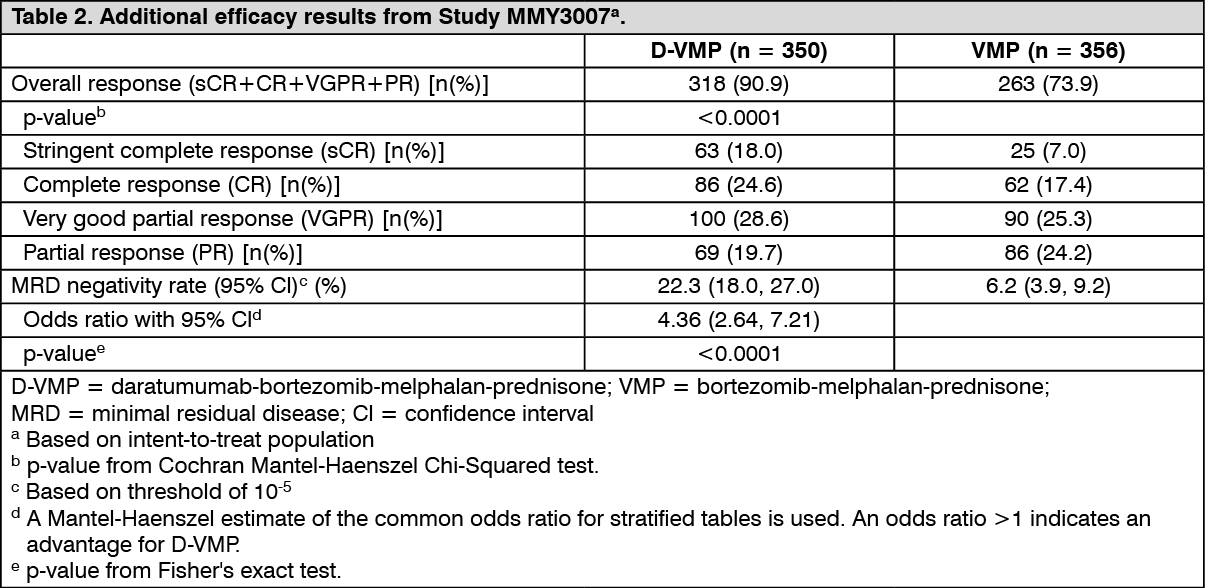 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn responders, the median time to response was 0.79 months (range: 0.4 to 15.5 months) in the D-VMP group and 0.82 months (range: 0.7 to 12.6 months) in the VMP group. The median duration of response had not been reached in the D-VMP group and was 21.3 months (range: 18.4, not estimable) in the VMP group.
A subgroup analysis was performed on patients at least 70 years old, or those 65-69 years old with ECOG performance score of 2, or aged less than 65 years of age with significant comorbidity or ECOG performance score of 2 (D-VMP: n=273, VMP: n=270). The efficacy results in this subgroup were consistent with the overall population. In this subgroup, median PFS was not reached in the D-VMP group and was 17.9 months in the VMP group (HR=0.56; 95% CI: 0.42, 0.75); p<0.0001). The overall response rate was 90% in the D-VMP group and 74% in the VMP group (VGPR rate: 29% in D-VMP group and 26% in VMP group; CR: 22% in D-VMP group and 18% in VMP group; sCR rate: 20% in D-VMP group and 7% in VMP group). The safety results of this subgroup were consistent with the overall population. Furthermore, safety analysis of the subgroup of patients with an ECOG performance score of 2 (D-VMP: n=89, VMP: n=84), was also consistent with the overall population.
Combination treatment with bortezomib, thalidomide and dexamethasone (VTd) in patients eligible for autologous stem cell transplant (ASCT): Study MMY3006 is a 2-part, open-label, randomised, active-controlled Phase III study. Part 1 compared induction and consolidation treatment with intravenous DARZALEX 16 mg/kg in combination with bortezomib, thalidomide and dexamethasone (D-VTd) to treatment with bortezomib, thalidomide and dexamethasone (VTd) in patients with newly diagnosed multiple myeloma eligible for ASCT. The consolidation phase of treatment began a minimum of 30 days post-ASCT, when the patient had recovered sufficiently, and engraftment was complete. In Part 2, subjects with at least a partial response (PR) by Day 100 post-transplant were re-randomised in a 1:1 ratio to daratumumab maintenance or observation only. Only results from Part 1 are described henceforth.
Bortezomib was administered by subcutaneous injection or intravenous injection at a dose of 1.3 mg/m2 body surface area twice weekly for two weeks (Days 1, 4, 8, and 11) of repeated 28 day (4-week) induction treatment cycles (Cycles 1-4) and two consolidation cycles (Cycles 5 and 6) following ASCT after Cycle 4. Thalidomide was administered orally at 100 mg daily during the six bortezomib cycles. Dexamethasone (oral or intravenous) was administered at 40 mg on Days 1, 2, 8, 9, 15, 16, 22 and 23 of Cycles 1 and 2, and at 40 mg on Days 1-2 and 20 mg on subsequent dosing days (Days 8, 9, 15, 16) of Cycles 3-4. Dexamethasone 20 mg was administered on Days 1, 2, 8, 9, 15, 16 in Cycles 5 and 6. On the days of intravenous DARZALEX infusion, the dexamethasone dose was administered intravenously as a pre-infusion medicinal product. Dose adjustments for bortezomib, thalidomide and dexamethasone were applied according to manufacturer's prescribing information.
A total of 1085 patients were randomised: 543 to the D-VTd arm and 542 to the VTd arm. The baseline demographic and disease characteristics were similar between the two treatment groups. The median age was 58 (range: 22 to 65) years. All patients were ≤65 years: 43% were in the age group ≥60-65 years, 41% were in the age group ≥50-60 years and 16% below age of 50 years. The majority were male (59%), 48% had an ECOG performance score of 0, 42% had an ECOG performance score of 1 and 10% had an ECOG performance score of 2. Forty percent had International Staging System (ISS) Stage I, 45% had ISS Stage II and 15% had ISS Stage III disease.
Efficacy was evaluated by the stringent Complete Response (sCR) rate at Day 100 post-transplant and Progression free survival (PFS). (See Table 3.)
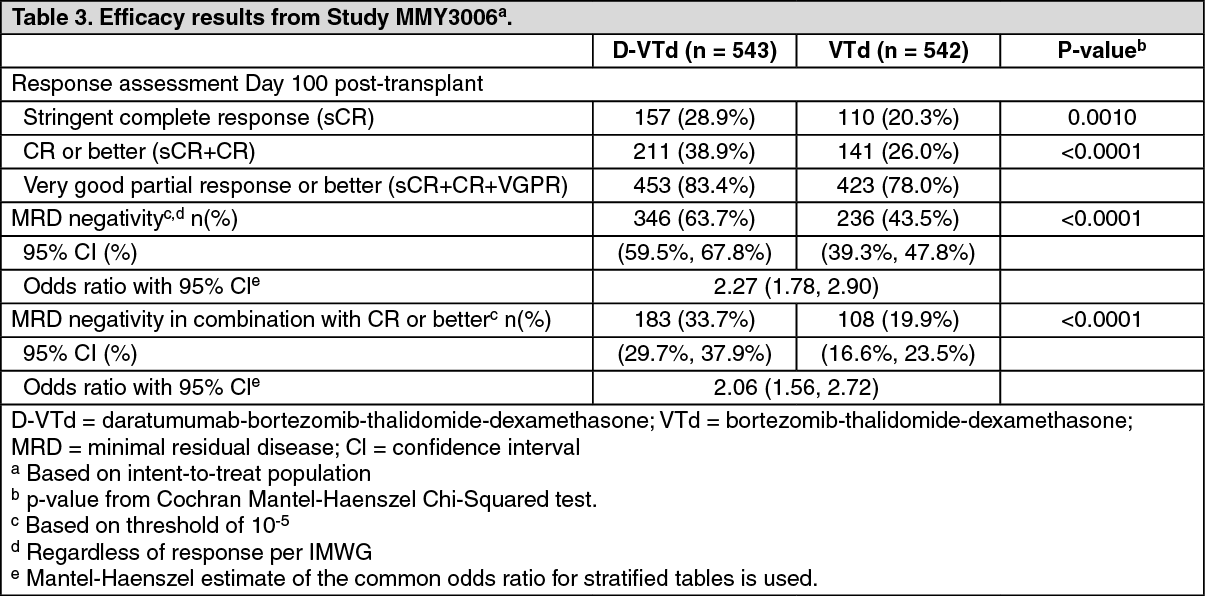 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageWith a median follow-up of 18.8 months, the primary analysis of PFS by censoring patients who were randomised to daratumumab maintenance in the second randomisation at the date of the second randomisation showed HR=0.50; 95% CI: 0.34, 0.75; p=0.0005. Results of an updated PFS analysis with a median follow-up of 44.5 months, censoring patients who were randomised to daratumumab maintenance in the second randomisation, showed HR=0.43; 95% CI: 0.33, 0.55; p < 0.0001. Median PFS was not reached in the D-VTd arm and was 37.8 months in the VTd arm. (See Figure 4.)
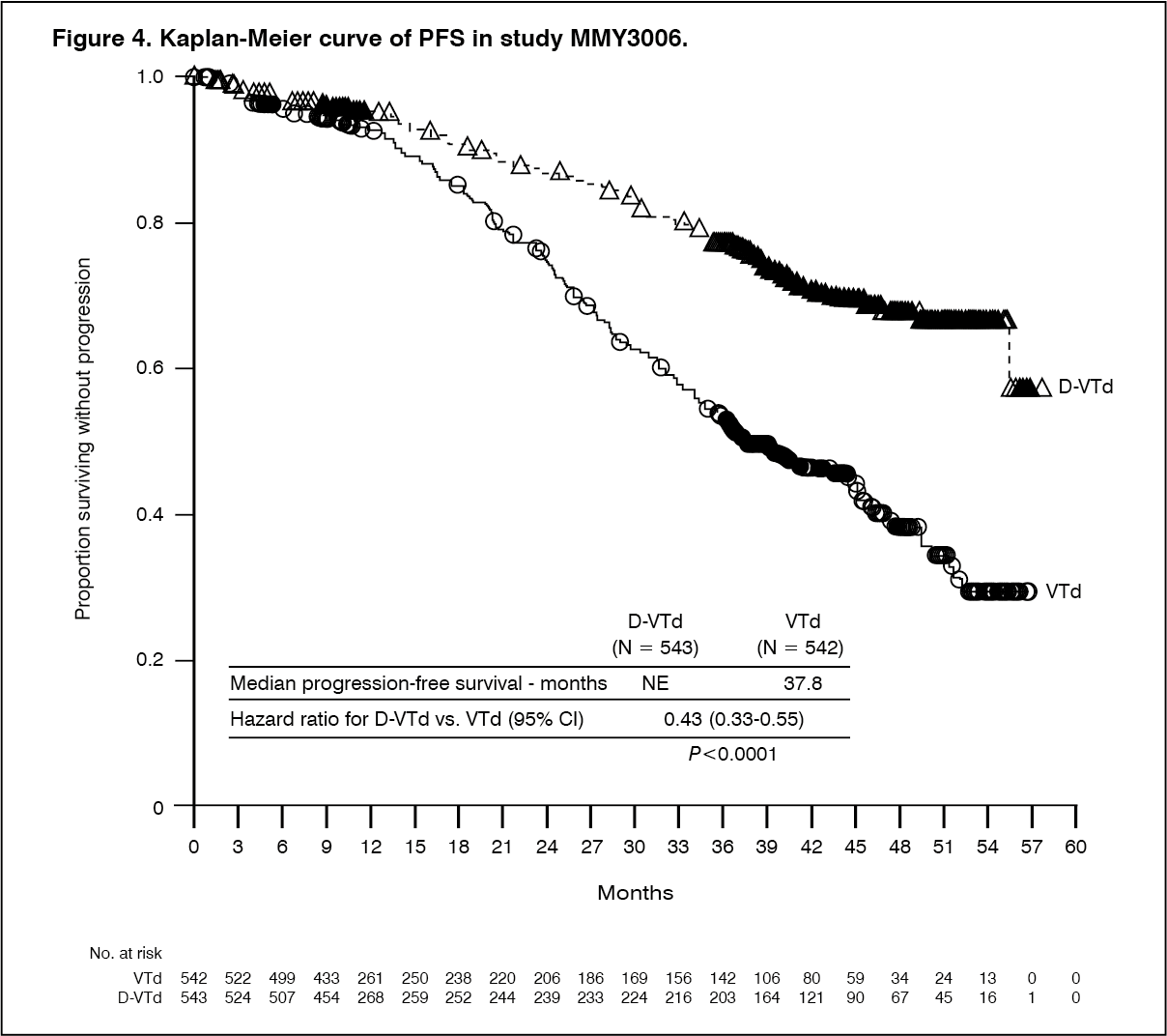 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageRelapsed/refractory multiple myeloma: Monotherapy: The clinical efficacy and safety of intravenous DARZALEX monotherapy for the treatment of adult patients with relapsed and refractory multiple myeloma whose prior therapy included a proteasome inhibitor and an immunomodulatory agent and who had demonstrated disease progression on the last therapy, was demonstrated in two open-label studies.
In Study MMY2002, 106 patients with relapsed and refractory multiple myeloma received 16 mg/kg intravenous DARZALEX until disease progression. The median patient age was 63.5 years (range, 31 to 84 years), 11% of patients were ≥ 75 years of age, 49% were male and 79% were Caucasian. Patients had received a median of 5 prior lines of therapy. Eighty percent of patients had received prior autologous stem cell transplantation (ASCT). Prior therapies included bortezomib (99%), lenalidomide (99%), pomalidomide (63%) and carfilzomib (50%). At baseline, 97% of patients were refractory to the last line of treatment, 95% were refractory to both, a proteasome inhibitor (PI) and immunomodulatory agent (IMiD), 77% were refractory to alkylating agents, 63% were refractory to pomalidomide and 48% of patients were refractory to carfilzomib.
Efficacy results of the pre-planned interim analysis based on Independent Review Committee (IRC) assessment are presented in Table 4 as follows. (See Table 4.)
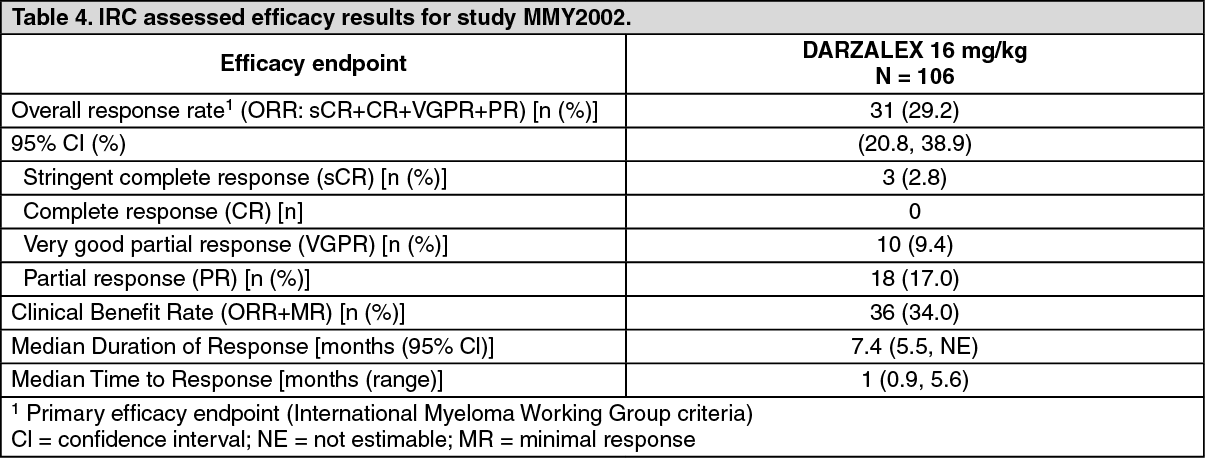 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageOverall response rate (ORR) in MMY2002 was similar regardless of type of prior anti-myeloma therapy. At a survival update with a median duration of follow-up of 14.7 months, median Overall Survival (OS) was 17.5 months (95% CI:13.7, not estimable).
In Study GEN501, 42 patients with relapsed and refractory multiple myeloma received 16 mg/kg intravenous DARZALEX until disease progression. The median patient age was 64 years (range, 44 to 76 years), 64% were male and 76% were Caucasian. Patients in the study had received a median of 4 prior lines of therapy. Seventy-four percent of patients had received prior ASCT. Prior therapies included bortezomib (100%), lenalidomide (95%), pomalidomide (36%) and carfilzomib (19%). At baseline, 76% of patients were refractory to the last line of treatment, 64% were refractory to both a PI and IMiD, 60% were refractory to alkylating agents, 36% were refractory to pomalidomide and 17% were refractory to carfilzomib.
Pre-planned interim analysis showed that treatment with daratumumab at 16 mg/kg led to a 36% ORR with 5% CR and 5% VGPR. The median time to response was 1 (range: 0.5 to 3.2) month. The median duration of response was not reached (95% CI: 5.6 months, not estimable).
At a survival update with a median duration of follow-up of 15.2 months, median OS was not reached (95% CI: 19.9 months, not estimable), with 74% of subjects still alive.
Combination treatment with lenalidomide: Study MMY3003, an open-label, randomised, active-controlled Phase III trial, compared treatment with intravenous DARZALEX 16 mg/kg in combination with lenalidomide and low-dose dexamethasone (DRd) to treatment with lenalidomide and low-dose dexamethasone (Rd) in patients with relapsed or refractory multiple myeloma who had received at least one prior therapy. Lenalidomide (25 mg once daily orally on Days 1-21 of repeated 28-day [4-week] cycles) was given with low dose dexamethasone at 40 mg/week (or a reduced dose of 20 mg/week for patients > 75 years or BMI < 18.5). On DARZALEX infusion days, 20 mg of the dexamethasone dose was given as a pre-infusion medicinal product and the remainder given the day after the infusion. Treatment was continued in both arms until disease progression or unacceptable toxicity.
A total of 569 patients were randomised; 286 to the DRd arm and 283 to the Rd arm. The baseline demographic and disease characteristics were similar between the intravenous DARZALEX and the control arm. The median patient age was 65 years (range 34 to 89 years) and 11% were ≥ 75 years. The majority of patients (86%) received a prior PI, 55% of patients had received a prior IMiD, including 18% of patients who had received prior lenalidomide; and 44% of patients had received both a prior PI and IMiD. At baseline, 27% of patients were refractory to the last line of treatment. Eighteen percent (18%) of patients were refractory to a PI only, and 21% were refractory to bortezomib. Patients refractory to lenalidomide were excluded from the study.
With a median follow‑up of 13.5 months, the primary analysis of PFS in study MMY3003 demonstrated an improvement in the DRd arm as compared to the Rd arm; the median PFS had not been reached in the DRd arm and was 18.4 months in the Rd arm (HR=0.37; 95% CI: 0.27, 0.52; p < 0.0001). Results of an updated PFS analysis after a median follow‑up of 55 months continued to show an improvement in PFS for patients in the DRd arm compared with the Rd arm. Median PFS was 45.0 months in the DRd arm and 17.5 months in the Rd arm (HR=0.44; 95% CI: 0.35, 0.54; p<0.0001), representing a 56% reduction in the risk of disease progression or death in patients treated with DRd (see Figure 5).
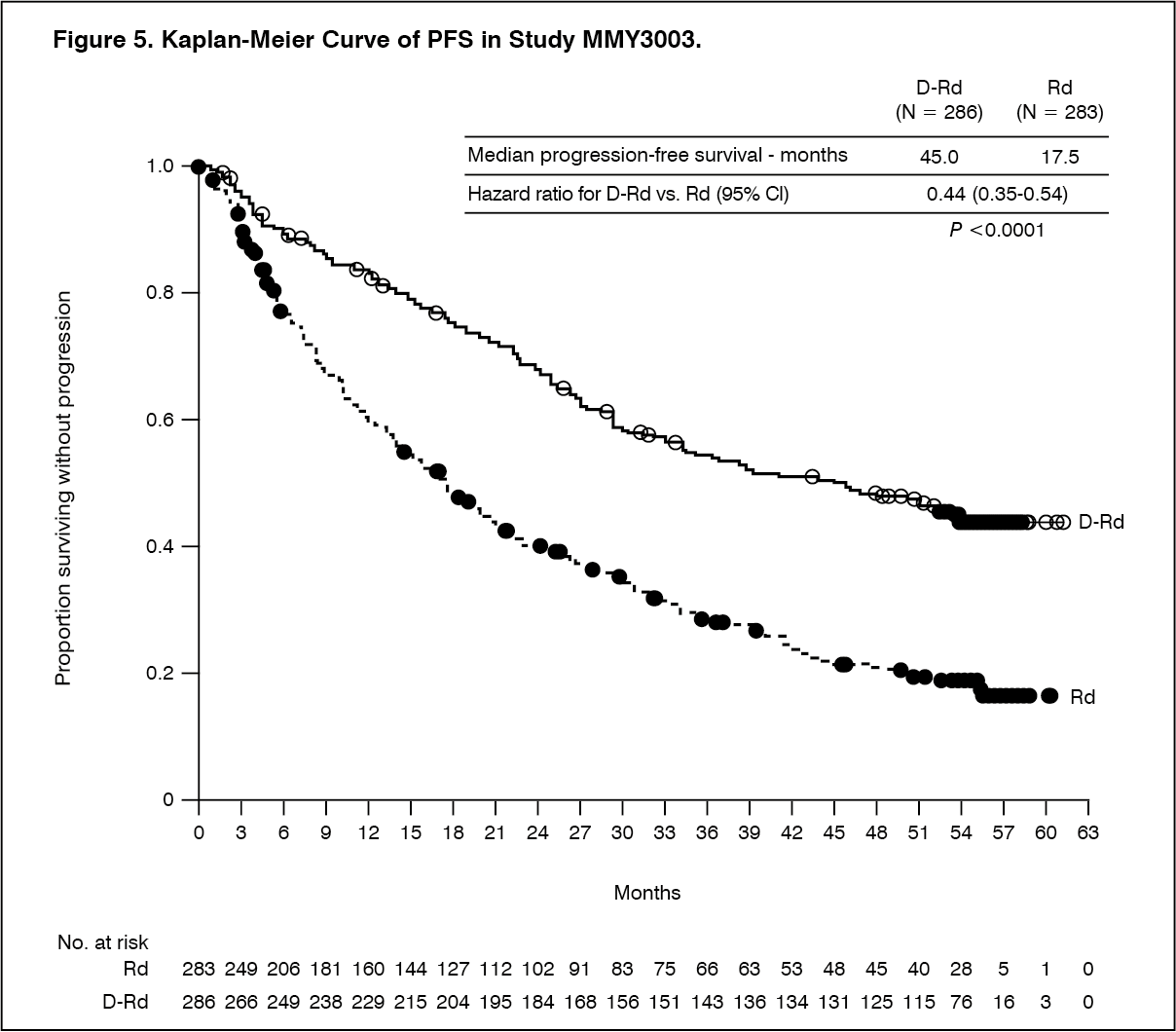 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAdditional efficacy results from Study MMY3003 are presented in Table 5 as follows. (See Table 5.)
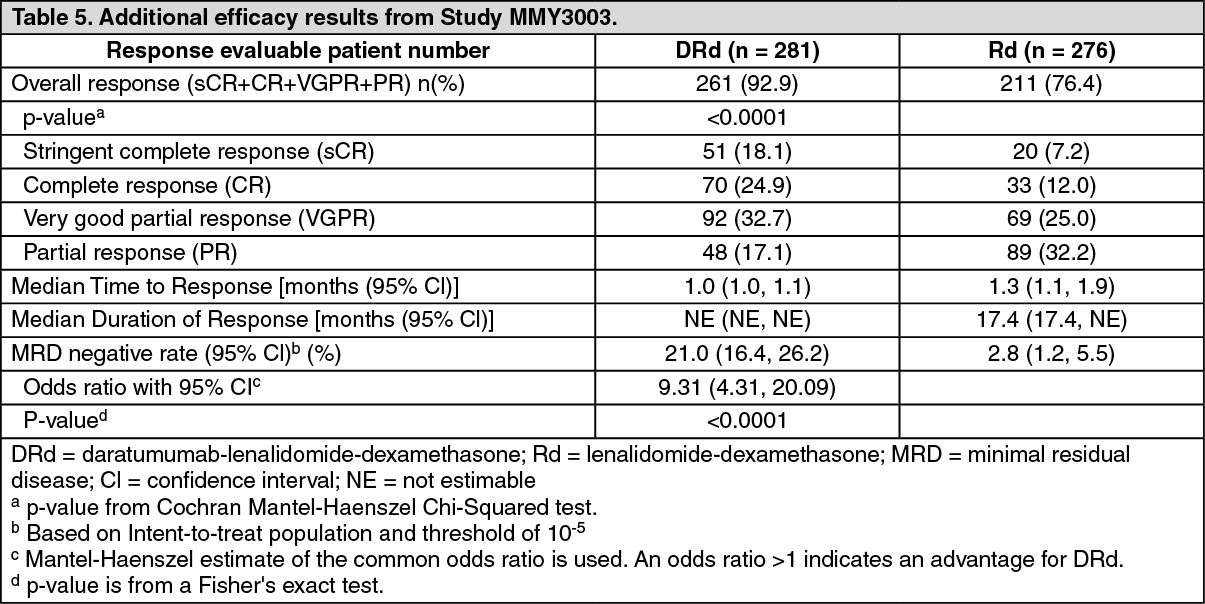 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageMedian OS was not reached for either treatment group. With an overall median follow-up of 13.5 months, the hazard ratio for OS was 0.64 (95% CI: 0.40, 1.01; p = 0.0534).
Combination treatment with bortezomib: Study MMY3004, an open-label, randomised, active-controlled Phase III trial, compared treatment with intravenous DARZALEX 16 mg/kg in combination with bortezomib and dexamethasone (DVd), to treatment with bortezomib and dexamethasone (Vd) in patients with relapsed or refractory multiple myeloma who had received at least one prior therapy. Bortezomib was administered by subcutaneous injection or intravenous infusion at a dose of 1.3 mg/m2 body surface area twice weekly for two weeks (Days 1, 4, 8, and 11) of repeated 21 day (3-week) treatment cycles, for a total of 8 cycles. Dexamethasone was administered orally at a dose of 20 mg on Days 1, 2, 4, 5, 8, 9, 11, and 12 of each of the 8 bortezomib cycles (80 mg/week for two out of three weeks of the bortezomib cycle) or a reduced dose of 20 mg/week for patients > 75 years, BMI < 18.5, poorly controlled diabetes mellitus or prior intolerance to steroid therapy. On the days of intravenous DARZALEX infusion, 20 mg of the dexamethasone dose was administered as a pre-infusion medicinal product. Intravenous DARZALEX treatment was continued until disease progression or unacceptable toxicity.
A total of 498 patients were randomised; 251 to the DVd arm and 247 to the Vd arm. The baseline demographic and disease characteristics were similar between the intravenous DARZALEX and the control arm. The median patient age was 64 years (range 30 to 88 years) and 12% were ≥ 75 years.
Sixty-nine percent (69%) of patients had received a prior PI (66% received bortezomib) and 76% of patients received an IMiD (42% received lenalidomide). At baseline, 32% of patients were refractory to the last line of treatment. Thirty-three percent (33%) of patients were refractory to an IMiD only, and 28% were refractory to lenalidomide. Patients refractory to bortezomib were excluded from the study.
With a median follow‑up of 7.4 months, the primary analysis of PFS in study MMY3004 demonstrated an improvement in the DVd arm as compared to the Vd arm; the median PFS had not been reached in the DVd arm and was 7.2 months in the Vd arm (HR [95% CI]: 0.39 [0.28, 0.53]; p-value < 0.0001). Results of an updated PFS analysis after a median follow‑up of 50 months continued to show an improvement in PFS for patients in the DVd arm compared with the Vd arm. Median PFS was 16.7 months in the DVd arm and 7.1 months in the Vd arm (HR [95% CI]: 0.31 [0.24, 0.39]; p-value <0.0001), representing a 69% reduction in the risk of disease progression or death in patients treated with DVd versus Vd (see Figure 6).
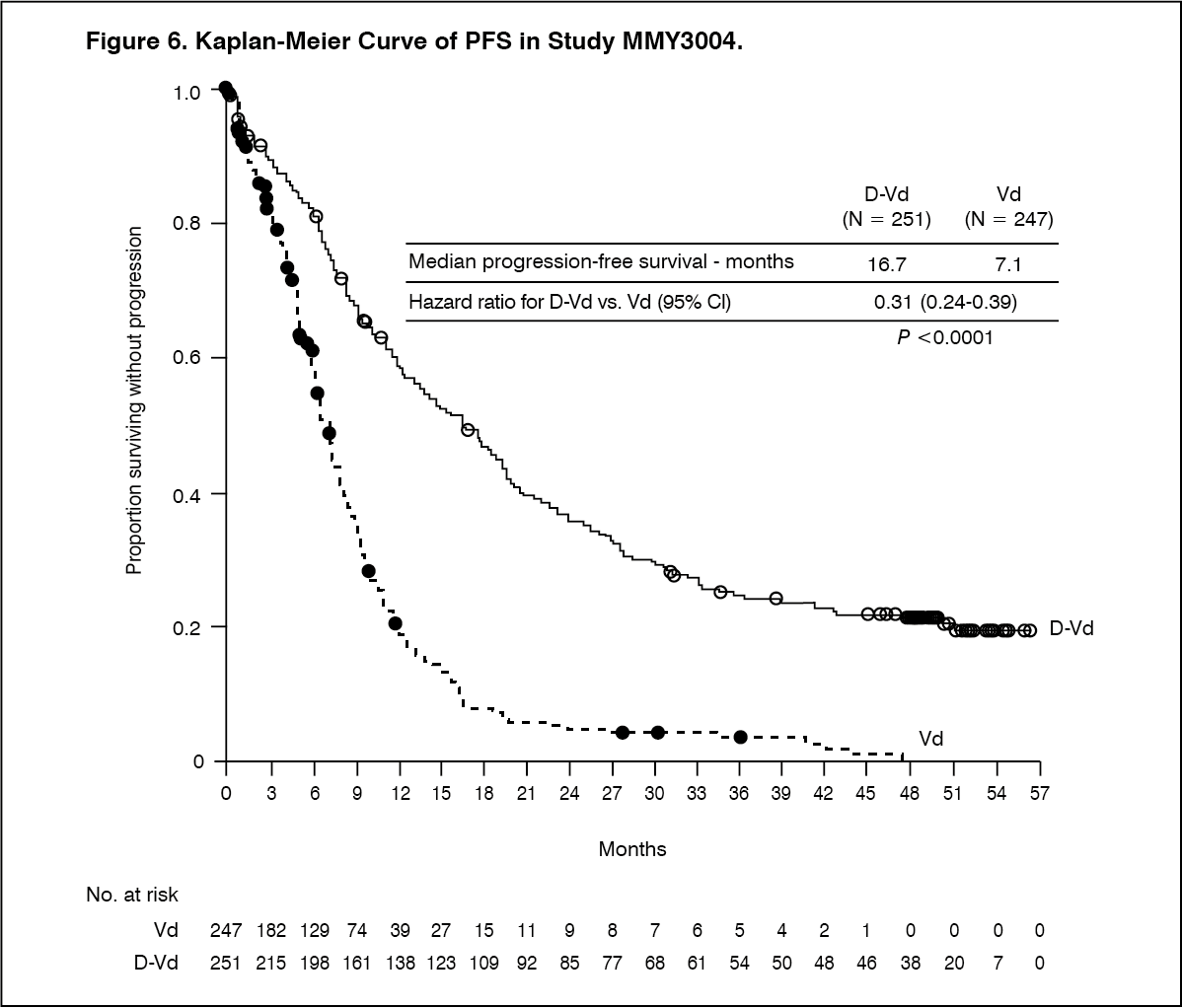 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAdditional efficacy results from Study MMY3004 are presented in Table 6 as follows. (See Table 6.)
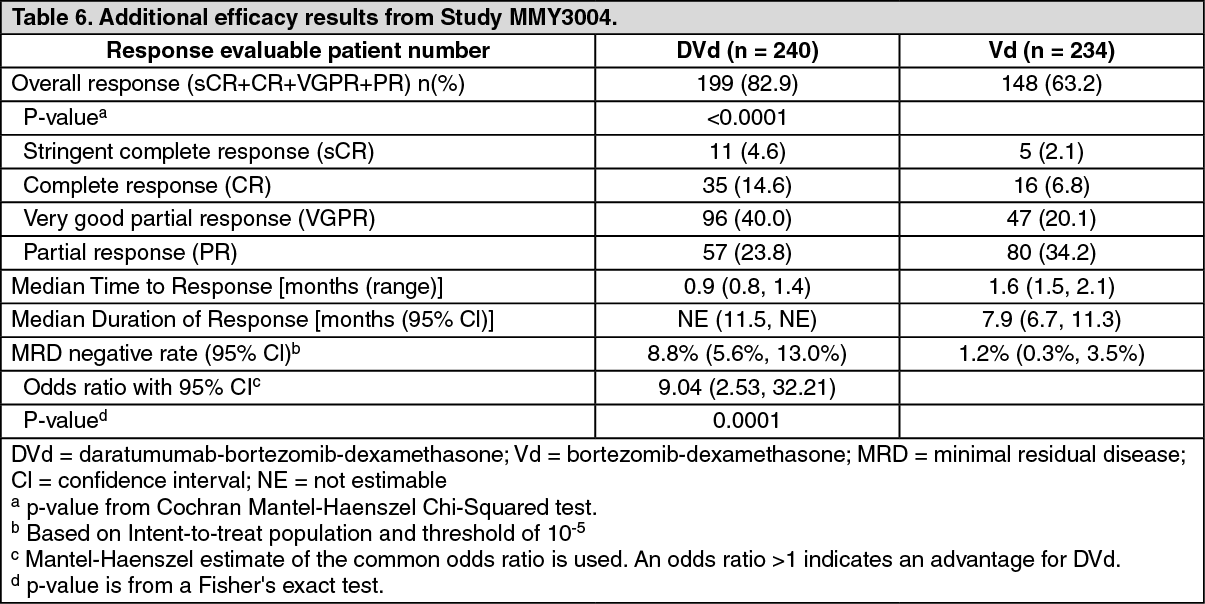 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageMedian OS was not reached for either treatment group. With an overall median follow-up of 7.4 months (95% CI: 0.0, 14.9), the hazard ratio for OS was 0.77 (95% CI: 0.47, 1.26; p = 0.2975).
Clinical experience of DARZALEX solution for subcutaneous injection (subcutaneous formulation): Monotherapy - relapsed/refractory multiple myeloma: MMY3012, an open-label, randomised, phase III non-inferiority study, compared efficacy and safety of treatment with DARZALEX solution for subcutaneous injection (1800 mg) vs. intravenous (16 mg/kg) daratumumab in patients with relapsed or refractory multiple myeloma who had received at least 3 prior lines of therapy including a proteasome inhibitor (PI) and an immunomodulatory agent (IMiD) or who were double-refractory to a PI and an IMiD. Treatment continued until unacceptable toxicity or disease progression.
A total of 522 patients were randomised: 263 to the DARZALEX subcutaneous formulation arm and 259 to the intravenous daratumumab arm. The baseline demographic and disease characteristics were similar between the two treatment groups. The median patient age was 67 years (range: 33 - 92 years), 55% were male and 78% were Caucasian. The median patient weight was 73 kg (range: 29 - 138 kg) Patients had received a median of 4 prior lines of therapy. A total of 51% of patients had prior autologous stem cell transplant (ASCT), 100% of patients were previously treated with both PI(s) and IMiD(s) and most patients were refractory to a prior systemic therapy, including both PI and IMiD (49%).
The study met its co-primary endpoints of overall response rate (ORR) by the IMWG response criteria (Table 7) and maximum Ctrough at pre-dose cycle 3 day 1, (see Dosage & Administration). (See Table 7.)
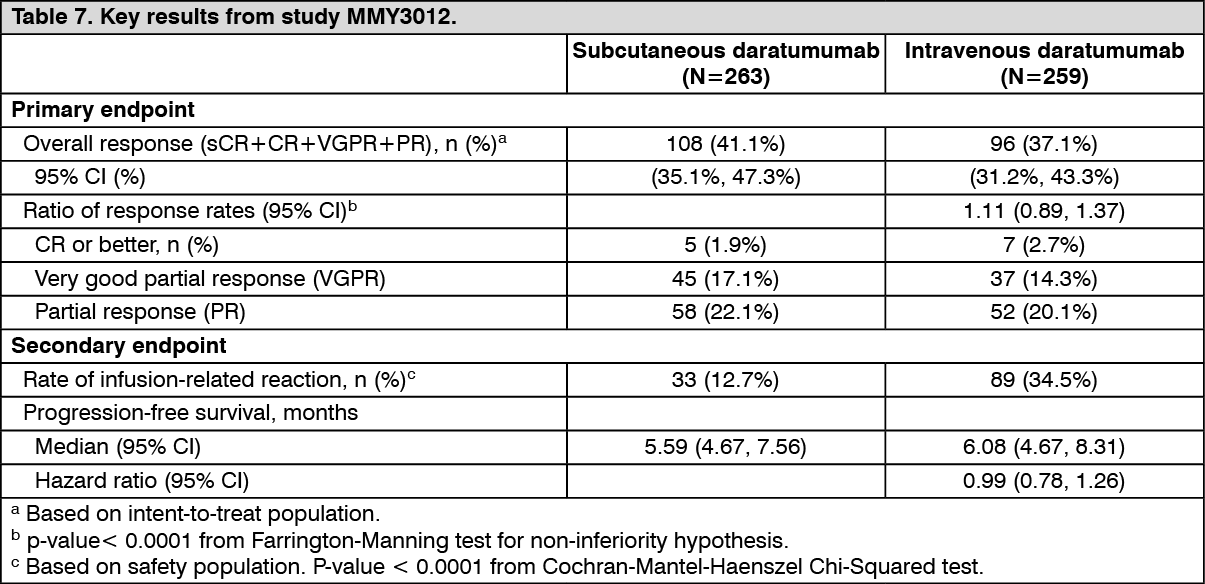 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAfter a median follow-up of 29.3 months, the median OS was 28.2 months (95% CI: 22.8, NE) in the DARZALEX subcutaneous formulation arm and was 25.6 months (95% CI: 22.1, NE) in the intravenous daratumumab arm.
Safety and tolerability results, including in lower weight patients, were consistent with the known safety profile for DARZALEX subcutaneous formulation and intravenous daratumumab.
Results from the modified-CTSQ, a patient reported outcome questionnaire that assesses patient satisfaction with their therapy, demonstrated that patients receiving DARZALEX subcutaneous formulation had greater satisfaction with their therapy compared with patients receiving intravenous daratumumab. However, open-label studies are subject to bias.
Combination therapies in multiple myeloma: MMY2040 was an open-label study evaluating the efficacy and safety of DARZALEX subcutaneous formulation 1800 mg: in combination with bortezomib, melphalan, and prednisone (D-VMP) in patients with newly diagnosed multiple myeloma (MM) who are ineligible for transplant. Bortezomib was administered by subcutaneous injection at a dose of 1.3 mg/m2 body surface area twice weekly at weeks 1, 2, 4 and 5 for the first 6-week cycle (cycle 1; 8 doses), followed by once weekly administrations at weeks 1, 2, 4 and 5 for eight more 6-week cycles (cycles 2-9; 4 doses per cycle). Melphalan at 9 mg/m2, and prednisone at 60 mg/m2 were orally administered on days 1 to 4 of the nine 6-week cycles (cycles 1-9). DARZALEX subcutaneous formulation was continued until disease progression or unacceptable toxicity.
In combination with lenalidomide and dexamethasone (D-Rd) in patients with relapsed or refractory MM. Lenalidomide (25 mg once daily orally on days 1-21 of repeated 28-day [4-week] cycles) was given with low dose dexamethasone 40 mg/week (or a reduced dose of 20 mg/week for patients > 75 years or BMI < 18.5). DARZALEX subcutaneous formulation was continued until disease progression or unacceptable toxicity.
In combination with bortezomib, lenalidomide, and dexamethasone (D-VRd) in patients with newly diagnosed MM who are transplant eligible. Bortezomib was administered by subcutaneous injection at a dose of 1.3 mg/m2 body surface area twice weekly at weeks 1 and 2. Lenalidomide was administered orally at 25 mg once daily on days 1-14; low dose dexamethasone was administered 40 mg/week in 3-week cycles. Total treatment duration was 4 cycles.
A total of 199 patients (D-VMP: 67; D-Rd: 65; D-VRd: 67) were enrolled. Efficacy results were determined by computer algorithm using IMWG criteria. The study met its primary endpoint ORR for D-VMP and D-Rd and the primary endpoint VGPR or better for D-VRd (see Table 8).
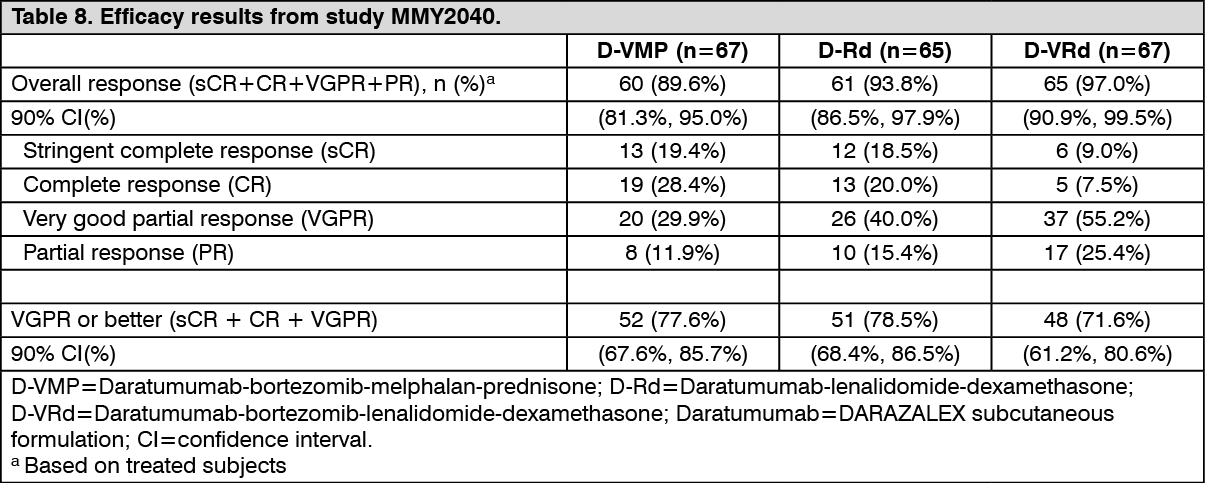 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageCombination treatment with pomalidomide and dexamethasone (Pd): Study MMY3013 was an open-label, randomised, active-controlled phase III study that compared treatment with DARZALEX subcutaneous formulation (1800 mg) in combination with pomalidomide and low-dose dexamethasone (D-Pd) to treatment with pomalidomide and low-dose dexamethasone (Pd) in patients with multiple myeloma who had received at least one prior line of therapy with lenalidomide and a proteasome inhibitor (PI). Pomalidomide (4 mg once daily orally on days 1-21 of repeated 28-day [4-week] cycles) was given with low dose oral or intravenous dexamethasone 40 mg/week (or a reduced dose of 20 mg/week for patients > 75 years). On DARZALEX subcutaneous formulation administration days, 20 mg of the dexamethasone dose was given as a pre-administration medicinal product and the remainder given the day after the administration. For patients on a reduced dexamethasone dose, the entire 20 mg dose was given as a DARZALEX subcutaneous formulation pre-administration medicinal product. Dose adjustments for pomalidomide and dexamethasone were applied according to manufacturer's prescribing information. Treatment was continued in both arms until disease progression or unacceptable toxicity.
A total of 304 patients were randomised: 151 to the D-Pd arm and 153 to the Pd arm. Patients with documented evidence of disease progression on or after the last regimen were included in the study. Patients who had ≥ grade 3 rash during prior therapy were excluded as per the pomalidomide Summary of Product Characteristics. The baseline demographic and disease characteristics were similar between the two treatment groups. The median patient age was 67 years (range 35 to 90 years), 18% were ≥ 75 years, 53% were male, and 89% Caucasian. Patients had received a median of 2 prior lines of therapy. All patients received a prior treatment with a proteasome inhibitor (PI) and lenalidomide, and 56% of patients received prior stem cell transplantation (ASCT). Ninety-six percent (96%) of patients received prior treatment with bortezomib. The majority of patients were refractory to lenalidomide (80%), a PI (48%), or both an immunomodulator and a PI (42%). Eleven percent of patients received 1 prior line of therapy; all were refractory to lenalidomide and 32.4% were refractory to both lenalidomide and a PI. Efficacy was evaluated by progression free survival (PFS) based on International Myeloma Working Group (IMWG) criteria.
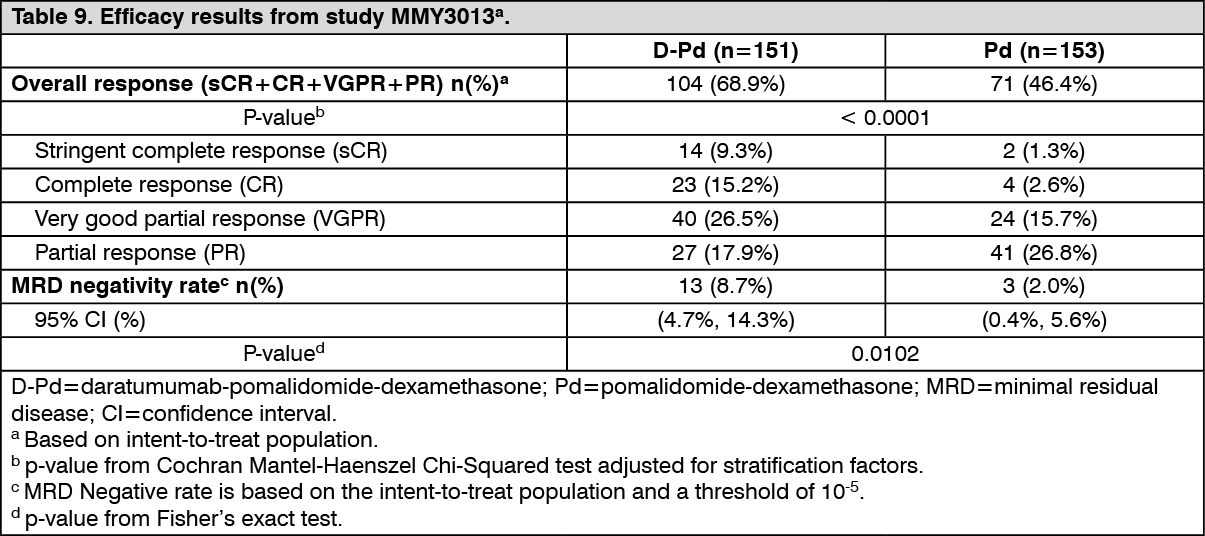
With a median follow-up of 16.9 months, the primary analysis of PFS in study MMY3013 showed a statistically significant improvement in the D-Pd arm as compared to the Pd arm; the median PFS was 12.4 months in the D-Pd arm and 6.9 months in the Pd arm (HR [95% CI]: 0.63 [0.47, 0.85]; p-value = 0.0018), representing a 37% reduction in the risk of disease progression or death for patients treated with D-Pd versus Pd. Median OS was not reached for either treatment group. (See Figure 7.)
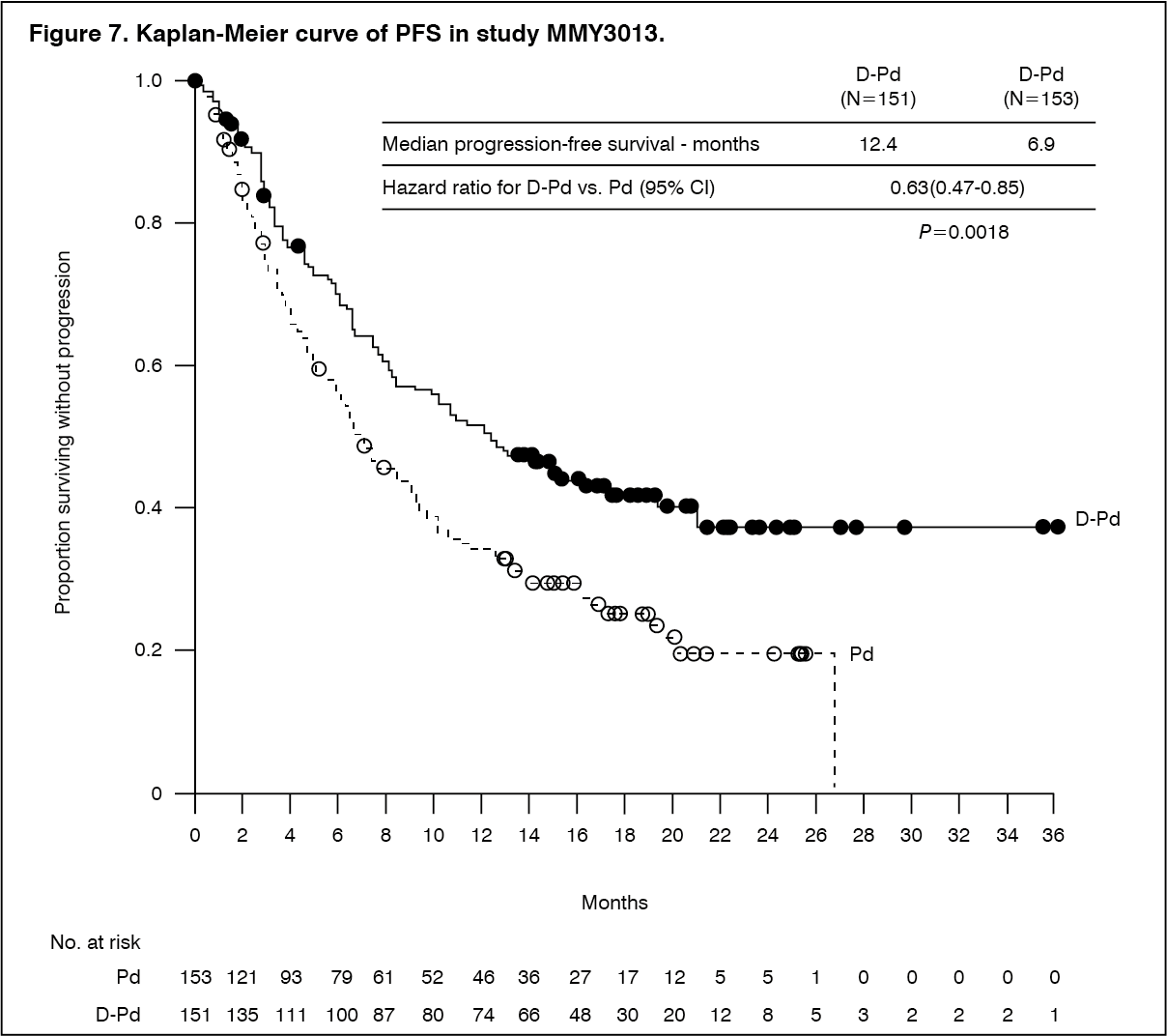 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAdditional efficacy results from study MMY3013 are presented in Table 9 as follows. (See Table 9.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn responders, the median time to response was 1 month (range: 0.9 to 9.1 months) in the D-Pd group and 1.9 months (range: 0.9 to 17.3 months) in the Pd group. The median duration of response had not been reached in the D-Pd group (range: 1 to 34.9+ months) and was 15.9 months (range: 1+ to 24.8 months) in the Pd group.
Combination treatment with bortezomib, cyclophosphamide and dexamethasone in patients with AL amyloidosis: Study AMY3001, an open-label, randomised, active-controlled phase III study, compared treatment with DARZALEX subcutaneous formulation (1800 mg) in combination with bortezomib, cyclophosphamide and dexamethasone (D-VCd) to treatment with bortezomib, cyclophosphamide and dexamethasone (VCd) alone in patients with newly diagnosed systemic AL amyloidosis. Randomisation was stratified by AL amyloidosis Cardiac Staging System, countries that typically offer autologous stem cell transplant (ASCT) for patients with AL amyloidosis, and renal function.
All patients enrolled in study AMY3001 had newly diagnosed AL amyloidosis with at least one affected organ, measurable hematologic disease, cardiac stage I-IIIA (based on European Modification of Mayo 2004 cardiac stage), and NYHA class I-IIIA. Patients with NYHA class IIIB and IV were excluded.
Bortezomib (SC; 1.3 mg/m2 body surface area), cyclophosphamide (oral or IV; 300 mg/m2 body surface area; max dose 500 mg), and dexamethasone (oral or IV; 40 mg or a reduced dose of 20 mg for patients > 70 years or body mass index [BMI] < 18.5 or those who have hypervolemia, poorly controlled diabetes mellitus or prior intolerance to steroid therapy) were administered weekly on days 1, 8, 15, and 22 of repeated 28-day [4-week] cycles. On the days of DARZALEX dosing, 20 mg of the dexamethasone dose was given as a pre-injection medicinal product and the remainder given the day after DARZALEX administration. Bortezomib, cyclophosphamide and dexamethasone were given for six 28-day [4-week] cycles in both treatment arms, while DARZALEX treatment was continued until disease progression, start of subsequent therapy, or a maximum of 24 cycles (~2 years) from the first dose of study treatment. Dose adjustments for bortezomib, cyclophosphamide and dexamethasone were applied according to manufacturer's prescribing information.
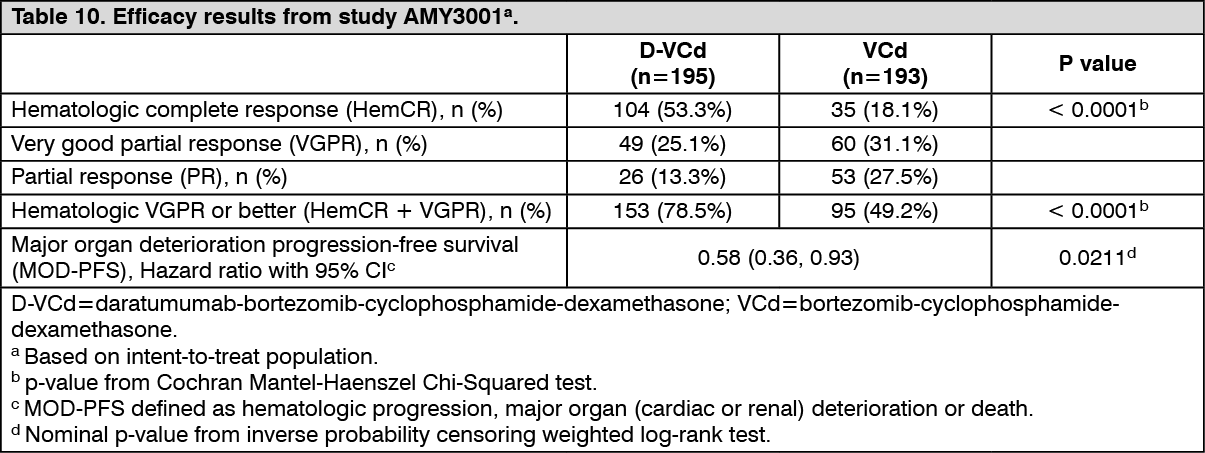
A total of 388 patients were randomised: 195 to the D-VCd arm and 193 to the VCd arm. The baseline demographic and disease characteristics were similar between the two treatment groups. The majority (79%) of patients had lambda free light chain disease. The median patient age was 64 years (range: 34 to 87); 47% were ≥ 65 years; 58% were male; 76% Caucasian, 17% Asian, and 3% African American; 23% had AL amyloidosis Clinical Cardiac stage I, 40% had stage II, 35% had stage IIIA, and 2% had stage IIIB. All patients had one or more affected organs and the median number of organs involved was 2 (range: 1-6) and 66% of patients had 2 or more organs involved. Vital organ involvement was: 71% cardiac, 59% renal and 8% hepatic. Patients with grade 2 sensory or grade 1 painful peripheral neuropathy were excluded. The primary efficacy endpoint was hematologic complete response (HemCR) rate as determined by the Independent Review Committee assessment based on International Concensus Criteria. Study AMY3001 demonstrated an improvement in HemCR in the D-VCd arm as compared to the VCd arm. Efficacy results are summarised in Table 10. (See Table 10.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn responders, the median time to HemCR was 60 days (range: 8 to 299 days) in the D-VCd group and 85 days (range: 14 to 340 days) in the VCd group. The median time to VGPR or better was 17 days (range: 5 to 336 days) in the D-VCd group and 25 days (range: 8 to 171 days) in the VCd group. The median duration of HemCR had not been reached in either arm.
The median follow-up for the study is 11.4 months. The median major organ deterioration progression-free survival (MOD-PFS) was not reached for patients in either arm.
Overall survival (OS) data were not mature. A total of 56 deaths were observed [n=27 (13.8%) D-VCd vs. n=29 (15%) VCd group].
Cardiac electrophysiology: Daratumumab as a large protein has a low likelihood of direct ion channel interactions. The effect of daratumumab on the QTc interval was evaluated in an open-label study for 83 patients (Study GEN501) with relapsed and refractory multiple myeloma following daratumumab infusions (4 to 24 mg/kg). Linear mixed PK-PD analyses indicated no large increase in mean QTcF interval (i.e. greater than 20 ms) at daratumumab Cmax.
Paediatric population: The European Medicines Agency has waived the obligation to submit the results of studies with DARZALEX in all subsets of the paediatric population in multiple myeloma (see Dosage & Administration for information on paediatric use).
Pharmacokinetics: Concentrate for solution for IV infusion: The pharmacokinetics (PK) of daratumumab following intravenous administration of daratumumab monotherapy were evaluated in patients with relapsed and refractory multiple myeloma at dose levels from 0.1 mg/kg to 24 mg/kg.
In the 1 to 24 mg/kg cohorts, peak serum concentrations (Cmax) after the first dose increased in approximate proportion to dose and volume of distribution was consistent with initial distribution into the plasma compartment. Following the last weekly infusion, Cmax increased in a greater than dose-proportional manner, consistent with target mediated drug disposition. Increases in AUC were more than dose-proportional and clearance (CL) decreased with increasing dose. These observations suggest CD38 may become saturated at higher doses, after which the impact of target binding clearance is minimised and the clearance of daratumumab approximates the linear clearance of endogenous IgG1. Clearance also decreased with multiple doses, which may be related to tumour burden decreases.
Terminal half-life increases with increasing dose and with repeated dosing. The mean (standard deviation [SD]) estimated terminal half-life of daratumumab following the first 16 mg/kg dose was 9 (4.3) days. The estimated terminal half-life of daratumumab following the last 16 mg/kg dose increased, but there are insufficient data for a reliable estimation. Based on population PK analysis, the mean (SD) half-life associated with non-specific linear elimination was approximately 18 (9) days; this is the terminal half-life that can be expected upon complete saturation of target mediated clearance and repeat dosing of daratumumab.
At the end of weekly dosing for the recommended monotherapy schedule and dose of 16 mg/kg, the mean (SD) serum Cmax value was 915 (410.3) micrograms/mL, approximately 2.9-fold higher than following the first infusion. The mean (SD) predose (trough) serum concentration at the end of weekly dosing was 573 (331.5) micrograms/mL.
Four population PK analyses were performed to describe the PK characteristics of daratumumab and to evaluate the influence of covariates on the disposition of daratumumab in patients with multiple myeloma; Analysis 1 (n=223) in patients receiving DARZALEX monotherapy while Analysis 2 (n=694), Analysis 3 (n=352) and Analysis 4 (n=355) were conducted in patients with multiple myeloma that received daratumumab combination therapies. Analysis 2 included 694 patients (n=326 for lenalidomide-dexamethasone; n=246 for bortezomib-dexamethasone; n=99 for pomalidomide-dexamethasone; n=11 for bortezomib-melphalan-prednisone; and n=12 for bortezomib-thalidomide-dexamethasone) and Analysis 3 included 352 patients (bortezomib-melphalan-prednisone) and Analysis 4 included 355 patients (lenalidomide-dexamethasone).
Based on the population PK analysis of daratumumab monotherapy (Analysis 1), daratumumab steady state is achieved approximately 5 months into the every 4-week dosing period (by the 21st infusion), and the mean (SD) ratio of Cmax at steady-state to Cmax after the first dose was 1.6 (0.5). The mean (SD) central volume of distribution is 56.98 (18.07) mL/kg.
Three additional population PK analyses (Analysis 2, Analysis 3 and Analysis 4) were conducted in patients with multiple myeloma that received daratumumab combination therapies. Daratumumab concentration-time profiles were similar following the monotherapy and combination therapies. The mean estimated terminal half-life associated with linear clearance in combination therapy was approximately 15-23 days.
Based on the population PK analyses (Analyses 1-4) body weight was identified as a statistically significant covariate for daratumumab clearance. Therefore, body weight based dosing is an appropriate dosing strategy for the multiple myeloma patients.
Simulation of daratumumab pharmacokinetics was conducted for all recommended dosing schedules in 1,309 patients with multiple myeloma. The simulation results confirmed that the split and single dosing for the first dose provide similar PK, with the exception of the PK profile in the first day of the treatment.
Special populations: Age and gender: Based on four individual population PK analyses (1-4) in patients receiving daratumumab monotherapy or various combination therapies (Analyses 1-4), age (range: 31-93 years) had no clinically important effect on the PK of daratumumab, and the exposure of daratumumab was similar between younger (aged < 65 years, n = 518) and older (aged ≥ 65 to <75 years, n = 761; aged ≥ 75 years, n = 334) patients.
Gender did not affect exposure of daratumumab to a clinically relevant degree in the population PK analyses.
Renal impairment: No formal studies of daratumumab in patients with renal impairment have been conducted. Four individual population PK analyses were performed based on pre-existing renal function data in patients receiving daratumumab monotherapy, or various combination therapies (Analyses 1-4), and included a total of 441 patients with normal renal function (creatinine clearance [CRCL] ≥ 90 mL/min), 621 with mild renal impairment (CRCL < 90 and ≥ 60 mL/min), 523 with moderate renal impairment (CRCL < 60 and ≥ 30 mL/min), and 27 with severe renal impairment or end stage renal disease (CRCL < 30 mL/min). No clinically important differences in exposure to daratumumab were observed between patients with renal impairment and those with normal renal function.
Hepatic impairment: No formal studies of daratumumab in patients with hepatic impairment have been conducted. Changes in hepatic function are unlikely to have any effect on the elimination of daratumumab since IgG1 molecules such as daratumumab are not metabolised through hepatic pathways.
Four individual population PK analyses were performed in patients receiving daratumumab monotherapy or various combination therapies (Analyses 1-4), and included a total of 1404 patients with normal hepatic function (total bilirubin [TB] and aspartate aminotransferase [AST] ≤ upper limit of normal [ULN]), 189 with mild hepatic impairment (TB 1.0 x to 1.5 x ULN or AST > ULN) and 8 patients with moderate (TB > 1.5 x to 3.0 x ULN; n=7), or severe (TB > 3.0 x ULN; n=1) hepatic impairment. No clinically important differences in the exposure to daratumumab were observed between patients with hepatic impairment and those with normal hepatic function.
Race: Based on four individual population PK analyses in patients receiving either daratumumab monotherapy, or various combination therapies (Analyses 1-4), the exposure to daratumumab was similar between white (n=1371) and non-white subjects (n=242).
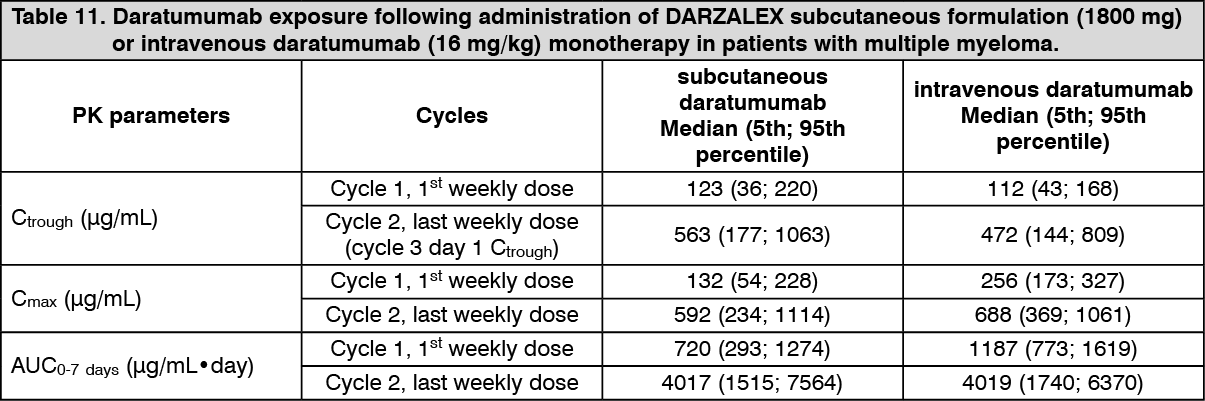
Solution for SC injection: In patients with multiple myeloma, daratumumab exposure in a monotherapy study following the recommended 1800 mg administration of DARZALEX subcutaneous formulation (weekly for 8 weeks, biweekly for 16 weeks, monthly thereafter) as compared to 16 mg/kg intravenous daratumumab for the same dosing schedule, showed non-inferiority for the co-primary endpoint of maximum Ctrough (cycle 3 day 1 pre-dose), with mean ± SD of 593 ± 306 μg/mL compared to 522 ± 226 μg/mL for intravenous daratumumab, with a geometric mean ratio of 107.93% (90% CI: 95.74-121.67).
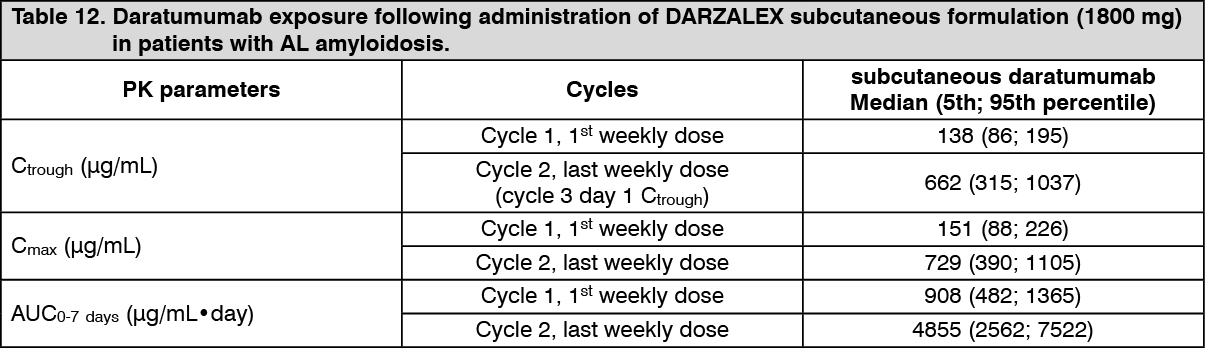
In a combination study, AMY3001, in patients with AL amyloidosis, the maximum Ctrough (cycle 3 day 1 pre-dose) was similar to that in multiple myeloma with mean ± SD of 597 ± 232 μg/mL following the recommended 1800 mg administration of DARZALEX subcutaneous formulation (weekly for 8 weeks, biweekly for 16 weeks, monthly thereafter).
Following the recommended dose of 1800 mg DARZALEX solution for subcutaneous injection, peak concentrations (Cmax) increased 4.8-fold and total exposure (AUC0-7 days) increased 5.4-fold from first dose to last weekly dose (8th dose). Highest trough concentrations for DARZALEX solution for subcutaneous injection are typically observed at the end of the weekly dosing regimens for both monotherapy and combination therapy.
In patients with multiple myeloma, the simulated trough concentrations following 6 weekly doses of 1800 mg DARZALEX solution for subcutaneous injection for combination therapy were similar to 1800 mg DARZALEX solution for subcutaneous injection monotherapy.
In patients with multiple myeloma, daratumumab exposure in a combination study with pomalidomide and dexamethasone (study MMY3013) was similar to that in monotherapy, with the maximum Ctrough (cycle 3 day 1 pre-dose) mean ± SD of 537 ± 277 μg/mL following the recommended 1800 mg administration of DARZALEX solution for subcutaneous injection (weekly for 8 weeks, biweekly for 16 weeks, monthly thereafter).
Absorption and distribution: At the recommended dose of 1800 mg in multiple myeloma patients, the absolute bioavailability of DARZALEX solution for subcutaneous injection is 69%, with an absorption rate of 0.012 hour-1, with peak concentrations occurring at 70 to 72 h (Tmax). At the recommended dose of 1800 mg in AL amyloidosis patients, the absolute bioavailability was not estimated, the absorption rate constant was 0.77 day-1 (8.31% CV) and peak concentrations occurred at 3 days.
The model predicted mean estimate of the volume of distribution for the central compartment was 5.25 L (36.9% CV) and peripheral compartment (V2) was 3.78 L in daratumumab monotherapy, and the modeled mean estimate of the volume of distribution for V1 was 4.36 L (28.0% CV) and V2 was 2.80 L when daratumumab was administered in combination with pomalidomide and dexamethasone in multiple myeloma patients. In AL amyloidosis patients, the model estimated apparent volume of distribution after subcutaneous administration is 10.8 L (3.1% CV). These results suggest that daratumumab is primarily localised to the vascular system with limited extravascular tissue distribution.
Metabolism and elimination: Daratumumab exhibits both concentration and time-dependent pharmacokinetics with parallel linear and nonlinear (saturable) elimination that is characteristic of target-mediated clearance. The population PK model estimated mean clearance value of daratumumab is 4.96 mL/h (58.7% CV) in daratumumab monotherapy and 4.32 mL/h (43.5% CV) when daratumumab is administered in combination with pomalidomide and dexamethasone in multiple myeloma patients. In AL amyloidosis patients, the apparent clearance after subcutaneous administration is 210 mL/day (4.1% CV). The model-based geometric mean for half-life associated with linear elimination is 20.4 days (22.4% CV) in daratumumab monotherapy and 19.7 days (15.3% CV) when daratumumab was administered in combination with pomalidomide and dexamethasone in multiple myeloma patients and 27.5 days (74.0% CV) in AL amyloidosis patients. For the monotherapy and combination regimens, the steady state is achieved at approximately 5 months into every 4 weeks dosage at the recommended dose and schedule (1800 mg; once weekly for 8 weeks, every 2 weeks for 16 weeks, and then every 4 weeks thereafter).
A population PK analysis was conducted using data from DARZALEX solution for subcutaneous injection monotherapy and combination therapy multiple myeloma studies, and the predicted PK exposures are summarised in Table 11. (See Table 11.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageA population PK analysis, using data from DARZALEX solution for subcutaneous injection combination therapy in AL amyloidosis patients, was conducted with data from 211 patients. At the recommended dose of 1800 mg, predicted daratumumab concentrations were slightly higher, but generally within the same range, in comparison with multiple myeloma patients. (See Table 12.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageSpecial populations: Age and gender: Based on population PK analyses in patients (33-92 years) receiving monotherapy or various combination therapies, age had no statistically significant effect on the PK of daratumumab. No individualisation is necessary for patients on the basis of age.
Gender had a statistically significant effect on PK parameters in patients with multiple myeloma but not in patients with AL amyloidosis. Slightly higher exposure in females were observed than males, but the difference in exposure is not considered clinically meaningful. No individualisation is necessary for patients on the basis of gender.
Renal impairment: No formal studies of DARZALEX subcutaneous formulation in patients with renal impairment have been conducted. Population PK analyses were performed based on pre-existing renal function data in patients with multiple myeloma receiving DARZALEX subcutaneous formulation monotherapy or various combination therapies in patients with multiple myeloma or AL amyloidosis. No clinically important differences in exposure to daratumumab were observed between patients with renal impairment and those with normal renal function.
Hepatic impairment: No formal studies of DARZALEX subcutaneous formulation in patients with hepatic impairment have been conducted.
Population PK analyses were performed in patients with multiple myeloma receiving DARZALEX subcutaneous formulation monotherapy or various combination therapies in patients with multiple myeloma and in AL amyloidosis. No clinically important differences in the exposure to daratumumab were observed between patients with normal hepatic function and mild hepatic impairment. There were very few patients with moderate and severe hepatic impairment to make meaningful conclusions for these populations.
Race: Based on the population PK analyses in patients receiving either DARZALEX subcutaneous formulation monotherapy or various combination therapies, the daratumumab exposure was similar across races.
Body weight: The flat-dose administration of DARZALEX subcutaneous formulation 1800 mg as monotherapy achieved adequate exposure for all body-weight subgroups. In patients with multiple myeloma, the mean cycle 3 day 1 Ctrough in the lower body-weight subgroup (≤ 65 kg) was 60% higher and in the higher body weight (> 85 kg) subgroup, 12% lower than the intravenous daratumumab subgroup. In some patients with body weight > 120 kg, lower exposure was observed which may result in reduced efficacy. However, this observation is based on limited number of patients.
In patients with AL amyloidosis, no meaningful differences were observed in Ctrough across body weight.
Toxicology: Preclinical safety data: Toxicology data have been derived from studies with daratumumab in chimpanzees and with a surrogate anti-CD38 antibody in cynomolgus monkeys. No chronic toxicity testing has been conducted.
Carcinogenicity and mutagenicity: No animal studies have been performed to establish the carcinogenic potential of daratumumab.
Reproductive toxicology: No animal studies have been performed to evaluate the potential effects of daratumumab on reproduction or development.
Fertility: No animal studies have been performed to determine potential effects on fertility in males or females.
Solution for SC injection: No carcinogenicity, genotoxicity, or fertility studies were conducted for recombinant human hyaluronidase. There were no effects on reproductive tissues and function and no systemic exposure of hyaluronidase in monkeys given 22000 U/kg/week subcutaneously (12 times higher than the human dose) for 39 weeks. As hyaluronidase is a recombinant form of the endogenous human hyaluronidase, no carcinogenicity, mutagenesis, or effects on fertility are expected.