Pharmacotherapeutic group: Drugs for obstructive airway diseases, other systemic drugs for obstructive airway diseases.
ATC code: R03DX09.
Pharmacology: Pharmacodynamics: Mechanism of action: Mepolizumab is a humanised monoclonal antibody (IgG1, kappa), which targets human interleukin-5 (IL-5) with high affinity and specificity. IL-5 is the major cytokine responsible for the growth and differentiation, recruitment, activation and survival of eosinophils. Mepolizumab inhibits the bioactivity of IL-5 with nanomolar potency by blocking the binding of IL-5 to the alpha chain of the IL-5 receptor complex expressed on the eosinophil cell surface, thereby inhibiting IL-5 signalling and reducing the production and survival of eosinophils.
Pharmacodynamic effects: Severe eosinophilic asthma: In patients with severe refractory eosinophilic asthma (adults/adolescents), following a dose of 100 mg administered subcutaneously every 4 weeks for 32 weeks, blood eosinophils were reduced from a geometric mean count at baseline of 290 to 40 cells/μL at week 32 (n=182), a reduction of 84% compared to placebo. This magnitude of blood eosinophils reduction was maintained in severe refractory eosinophilic asthma patients (n=998) treated for a median of 2.8 years (range 4 weeks to 4.5 years) in open-label extension studies.
In adults, adolescents and children, this magnitude of reduction was observed within 4 weeks of treatment.
CRSwNP: In patients with CRSwNP, following a 100 mg dose of mepolizumab administered subcutaneously every 4 weeks for 52 weeks, blood eosinophils were reduced from a geometric mean count at baseline to week 52 of 390 (n=206) to 60 cells/μL (n=126), which corresponds to a geometric mean reduction of 83% compared to placebo. This magnitude of reduction was observed within 4 weeks of treatment and was maintained throughout the treatment period of 52 weeks.
EGPA: In patients with EGPA, following a 300 mg dose of mepolizumab administered subcutaneously every 4 weeks for 52 weeks, blood eosinophils were reduced from a geometric mean count at baseline of 177 (n=68) to 38 cells/μL (n=64) at week 52. There was a geometric mean reduction of 83% compared to placebo and this magnitude of reduction was observed within 4 weeks of treatment.
HES: In patients with HES (adults/adolescents), following a 300 mg dose of mepolizumab administered subcutaneously every 4 weeks for 32 weeks, blood eosinophil reduction was observed within 2 weeks of treatment. At week 32, blood eosinophils were reduced from a geometric mean count at baseline of 1460 (n=54) to 70 cells/μL (n=48) and a geometric mean reduction of 92% compared to placebo was observed. This magnitude of reduction was maintained for a further 20 weeks in patients that continued mepolizumab treatment in the open-label extension study.
Immunogenicity: Severe eosinophilic asthma, CRSwNP, EGPA and HES: Consistent with the potentially immunogenic properties of protein and peptide therapeutics, patients may develop antibodies to mepolizumab following treatment. In the placebo-controlled trials, 15/260 (6%) of adults and adolescents with severe refractory eosinophilic asthma treated with 100 mg dose, 6/196 (3%) of adults with CRSwNP treated with 100 mg dose, 1/68 (<2%) of adults with EGPA treated with 300 mg dose and 1/53 (2%) of adults and adolescents with HES treated with 300 mg dose of mepolizumab subcutaneously had detectable anti-mepolizumab antibodies after having received at least one dose of mepolizumab.
The immunogenicity profile of mepolizumab in severe refractory eosinophilic asthma patients (n=998) treated for a median of 2.8 years (range 4 weeks to 4.5 years) or in HES patients (n=102) treated for 20 weeks in open-label extension studies was similar to that observed in the placebo-controlled studies.
Neutralising antibodies were detected in one adult patient with severe refractory eosinophilic asthma and in no patients with CRSwNP, EGPA or HES. Anti-mepolizumab antibodies did not discernibly impact the pharmacokinetics and pharmacodynamics of mepolizumab in the majority of patients and there was no evidence of a correlation between antibody titres and change in blood eosinophil level.
Clinical efficacy: Severe eosinophilic asthma: The efficacy of mepolizumab in the treatment of a targeted group of patients with severe refractory eosinophilic asthma was evaluated in 3 randomised, double-blind, parallel-group clinical studies of between 24-52 weeks duration, in patients aged 12 years and older. These patients either remained uncontrolled (at least two severe exacerbations in the previous 12 months) on their current standard of care, including at least high doses of inhaled corticosteroids (ICS) plus an additional maintenance treatment(s), or were dependent on systemic corticosteroids. Additional maintenance treatments included long-acting beta
2-adrenergic agonists (LABA), leukotriene modifiers, long-acting muscarinic antagonists (LAMA), theophylline, and oral corticosteroids (OCS).
The two exacerbations studies MEA112997 and MEA115588 enrolled a total of 1192 patients, 60% females, with a mean age of 49 years (range 12-82). The proportion of patients on maintenance OCS was 31% and 24%, respectively. Patients were required to have a history of two or more severe asthma exacerbations requiring oral or systemic corticosteroid treatment in the past 12 months and reduced lung function at baseline (pre-bronchodilator FEV
1 <80% in adults and <90% in adolescents). The mean number of exacerbations in the previous year was 3.6 and the mean predicted pre-bronchodilator FEV
1 was 60%. Patients continued to receive their existing asthma medicine during the studies.
For the oral corticosteroid-sparing study MEA115575, a total of 135 patients were enrolled (55% were female; mean age of 50 years) who were being treated daily with OCS (5-35 mg per day), and high-dose ICS plus an additional maintenance medicine.
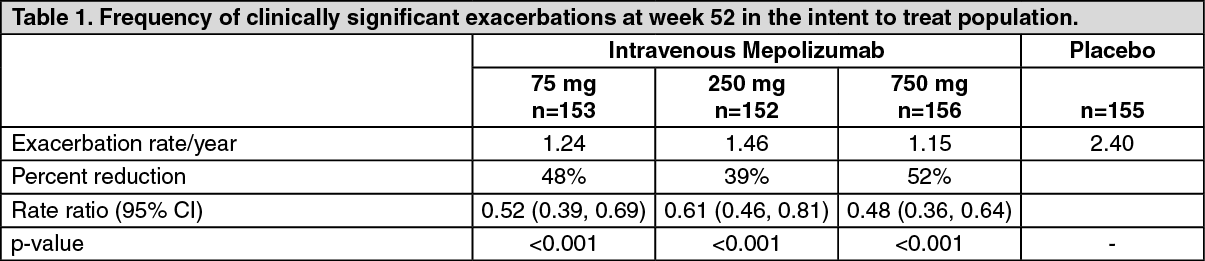
Dose-ranging efficacy MEA112997 (DREAM) study: In MEA112997, a randomised, double-blind, placebo-controlled, parallel-group, multi-centre study of 52 weeks duration in 616 patients with severe refractory eosinophilic asthma, mepolizumab significantly reduced clinically significant asthma exacerbations (defined as worsening of asthma requiring use of oral/systemic corticosteroids and/or hospitalisation and/or emergency department visits) when administered in doses of 75 mg, 250 mg or 750 mg intravenously compared to placebo (see Table 1).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Exacerbation reduction MEA115588 (MENSA) study: MEA115588 was a randomised, double-blind, placebo-controlled, parallel-group, multi-centre study which evaluated the efficacy and safety of mepolizumab as add-on therapy in 576 patients with severe refractory eosinophilic asthma defined as peripheral blood eosinophils greater than or equal to 150 cells/μL at initiation of treatment or greater than or equal to 300 cells/μL within the past 12 months.
Patients received mepolizumab 100 mg administered subcutaneously, mepolizumab 75 mg administered intravenously or placebo treatment once every 4 weeks over 32 weeks. The primary endpoint was the frequency of clinically significant exacerbations of asthma and the reductions for both mepolizumab treatment arms compared to placebo were statistically significant (p<0.001). Table 2 provides the results of the primary and secondary endpoints for patients treated with subcutaneous mepolizumab or placebo. (See Table 2.)
Click on icon to see table/diagram/image
Reduction of exacerbation rate by baseline blood eosinophil count: Table 3 shows the results of a combined analysis of the two exacerbation studies (MEA112997 and MEA115588) by baseline blood eosinophil count. The rate of exacerbations in the placebo arm increased with increasing baseline blood eosinophil count. The reduction rate with mepolizumab was greater in patients with higher blood eosinophil counts. (See Table 3.)
Click on icon to see table/diagram/image
Oral corticosteroid reduction study MEA115575 (SIRIUS): MEA115575 evaluated the effect of mepolizumab 100 mg administered subcutaneously on reducing the requirement for maintenance oral corticosteroids (OCS) while maintaining asthma control in subjects with severe refractory eosinophilic asthma. Patients had a blood eosinophil count of ≥150/μL at baseline or a blood eosinophil count of ≥300/μL in the 12 months prior to screening. Patients were administered mepolizumab or placebo treatment once every 4 weeks over the treatment period. Patients continued to receive their existing asthma medicine during the study with the exception of their OCS dose which was reduced every 4 weeks during the OCS reduction phase (Weeks 4-20), as long as asthma control was maintained.
A total of 135 patients were enrolled: mean age was 50 years, 55% were female, and 48% had been receiving oral steroid therapy for at least 5 years. The baseline mean prednisone equivalent dose was approximately 13 mg per day.
The primary endpoint was the percent reduction in daily OCS dose (weeks 20-24), whilst maintaining asthma control by defined dose reduction categories (see Table 4). Predefined categories included percent reductions ranging from 90-100% reduction, to no decrease in the prednisone dose from the end of the optimisation phase. The comparison between mepolizumab and placebo was statistically significant (p=0.008). (See Table 4.)
Click on icon to see table/diagram/image
Open-label extension studies in severe refractory eosinophilic asthma MEA115666 (COLUMBA), MEA115661 (COSMOS) and 201312 (COSMEX): The long-term efficacy profile of mepolizumab in severe refractory eosinophilic asthma patients (n=998) treated for a median of 2.8 years (range 4 weeks to 4.5 years) in open-label extension studies MEA115666, MEA115661 and 201312 was generally consistent with the 3 placebo-controlled studies.
Chronic Rhinosinusitis with Nasal Polyps (CRSwNP): Study 205687 (SYNAPSE) was a 52-week, randomised, double-blind, placebo-controlled study which evaluated 407 patients aged 18 years and older with CRSwNP.
Patients enrolled in the study were required to have a nasal obstruction VAS (Visual Analogue Scale) symptom score of >5 out of a maximum score of 10, an overall VAS symptom score >7 out of a maximum score of 10 and an endoscopic bilateral NP score of ≥5 out of a maximum score of 8 (with a minimum score of 2 in each nasal cavity). Patients must also have had a history of at least one prior surgery for nasal polyps in the previous 10 years.
Key baseline characteristics included total endoscopic NP score mean (SD) 5.5 (1.29), nasal obstruction VAS score mean (SD) 9.0 (0.83), overall VAS symptom score mean (SD) 9.1 (0.74), loss of smell VAS score mean (SD) 9.7 (0.72) and Sino-Nasal Outcome Test (SNOT-22) mean (SD) 64.1 (18.32). The geometric mean eosinophil count was 390 cells/mcL (95% CI: 360, 420). In addition, 27% of patients had aspirin-exacerbated respiratory disease (AERD) and 48% of patients had at least 1 course of OCS for CRSwNP in the past 12 months.
Patients received a 100 mg dose of mepolizumab or placebo, administered subcutaneously once every 4 weeks in addition to background intranasal corticosteroid therapy.
The co-primary endpoints were change from baseline in total endoscopic NP score at week 52 and change from baseline in mean nasal obstruction VAS score during weeks 49-52. The key secondary endpoint was the time to first NP surgery up to Week 52 (surgery was defined as any procedure involving instruments resulting in incision and removal of tissue [e.g. polypectomy] in the nasal cavity). Patients who received mepolizumab had significantly greater improvements (decreases) in total endoscopic NP score at Week 52 and in nasal obstruction VAS score during weeks 49-52 compared to placebo, and all secondary endpoints were statistically significant in favour of mepolizumab (see Table 5 and Figure 1).
Click on icon to see table/diagram/image
Time to First NP surgery: Across the 52-week treatment period, patients in the mepolizumab group had a lower probability of undergoing NP surgery than patients in the placebo group. The risk of surgery over the treatment period was significantly lower by 57% for patients treated with mepolizumab compared with placebo (Hazard Ratio: 0.43; 95% CI 0.25, 0.76; p=0.003).
Click on icon to see table/diagram/image
A post-hoc analysis of the proportion of patients with surgery showed a 61% reduction in the odds of surgery versus placebo (OR: 0.39, 95% CI: 0.21, 0.72; p=0.003).
CRSwNP patients with co-morbid asthma: In 289 (71%) patients with co-morbid asthma, pre-specified analyses showed improvements in the co-primary endpoints consistent with those seen in the overall population in the patients who received mepolizumab 100 mg compared with placebo. Additionally in these patients, there was a greater improvement from baseline at Week 52 in asthma control as measured by the Asthma Control Questionnaire (ACQ-5) for mepolizumab 100 mg compared with placebo (median change [Q1, Q3] of -0.80 [-2.20, 0.00] and 0.00 [-1.10, 0.20], respectively).
Eosinophilic Granulomatosis with Polyangiitis (EGPA): MEA115921 was a randomised, double-blind, placebo-controlled, 52-week study which evaluated 136 adult patients with EGPA, who had a history of relapsing or refractory disease, and who were on stable oral corticosteroid therapy (OCS; ≥7.5 to ≤50 mg/day prednisolone/prednisone), with or without stable immunosuppressant therapy (excluding cyclophosphamide). Other background standard of care therapy was allowed during the study. Fifty-three percent (n=72) were also on concomitant stable immunosuppressant therapy. Patients with organ-threatening or life-threatening EGPA were excluded from study MEA115921.
Patients either received a 300 mg dose of mepolizumab or placebo administered subcutaneously once every 4 weeks in addition to their background prednisolone/prednisone with or without immunosuppressive therapy. The OCS dose was tapered at the discretion of the investigator.
Remission: The co-primary endpoints were the total accrued duration of remission, defined as a Birmingham Vasculitis Activity Score (BVAS)=0 plus prednisolone/prednisone dose ≤4 mg/day, and the proportion of patients in remission at both 36 and 48 weeks of treatment. BVAS=0 represents no active vasculitis.
Compared with placebo, patients receiving mepolizumab 300 mg achieved a significantly greater accrued time in remission. Additionally, compared to placebo, a significantly higher proportion of patients receiving mepolizumab 300 mg achieved remission at both Week 36 and Week 48 (Table 6).
For both co-primary endpoints, compared with placebo, the beneficial effect observed following mepolizumab 300 mg treatment was present irrespective of if patients were receiving immunosuppressant therapy in addition to background corticosteroids.
Using the secondary endpoint remission definition of BVAS=0 plus prednisolone/prednisone ≤7.5 mg/day, patients receiving mepolizumab 300 mg also achieved significantly greater accrued time in remission (p<0.001), and a higher proportion of patients were in remission at both Week 36 and Week 48 (p<0.001), compared to placebo. (See Table 6.)
Click on icon to see table/diagram/image
Relapse: Compared with placebo, the time to first relapse was significantly longer for patients receiving mepolizumab 300 mg (p<0.001). Additionally, patients receiving mepolizumab had a 50% reduction in annualised relapse rate compared with placebo: 1.14 vs 2.27, respectively.
Oral corticosteroid reduction: Patients treated with mepolizumab had a significantly lower average daily OCS during Weeks 48-52 compared with patients who received placebo. During Weeks 48 to 52, 59% and 44% of patients treated with mepolizumab achieved an average daily OCS dose of ≤7.5 mg and ≤4 mg, respectively, compared with 33% and 7% in the placebo group. 18% of patients in the mepolizumab group were able to taper off OCS completely compared with 3% in the placebo group.
Asthma Control Questionnaire - 6 (ACQ-6): Patients treated with mepolizumab had significant improvements in mean ACQ-6 score during Weeks 49-52 compared with patients who received placebo.
Hypereosinophilic syndrome (HES): Study 200622 was a randomised, double-blind, placebo-controlled, 32-week study which evaluated 108 patients ≥12 years old with HES. Patients received 300 mg of mepolizumab, or placebo administered subcutaneously once every 4 weeks while continuing their HES therapy. In study 200622, HES therapy included but was not limited to OCS, immunosuppressive, cytotoxic therapy or other symptomatic therapies associated with HES such as omeprazole.
Patients entering the study had experienced at least two HES flares within the past 12 months and had a blood eosinophil count ≥1000 cells/μL during screening. Patients who were FIP1L1-PDGFRα kinase-positive were excluded from the study.
The primary endpoint of study 200622 was the proportion of patients who experienced a HES flare during the 32-week treatment period. A HES flare was defined as worsening of clinical signs and symptoms of HES resulting in the need to increase OCS or increase/add cytotoxic or immunosuppressive HES therapy or receiving blinded active OCS due to increased blood eosinophils (on ≥2 occasions).
The primary analysis compared patients who experienced a HES flare or withdrew from the study in the mepolizumab and placebo treatment groups. Over the 32-week treatment period, 50% fewer patients experienced a HES flare or withdrew from the study when treated with 300 mg mepolizumab compared with placebo; 28% versus 56%, respectively (OR 0.28, 95% CI: 0.12, 0.64) (see Table 7).
Secondary endpoints were time to first HES flare, proportion of patients who experienced a HES flare during Week 20 through Week 32, rate of HES flares and change from baseline in fatigue severity. All secondary endpoints were statistically significant and provided support for the primary endpoint (see Figure 2 and Table 8).
Click on icon to see table/diagram/image
Time to First Flare: Patients who received 300 mg mepolizumab had a significant increase in the time to first HES flare compared with placebo. The risk of first HES flare over the treatment period was 66% lower for patients treated with Nucala compared with placebo (Hazard Ratio: 0.34; 95% CI 0.18, 0.67; p=0.002).
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Open-label extension (OLE): Study 205203 was a 20-week open-label extension of Study 200622. HES therapy was allowed to be adjusted per local standard of care while maintaining mepolizumab 300 mg treatment starting at Week 4. In this study, the effect of treatment with mepolizumab on the reduction of HES flares reported during Study 200622 was sustained for patients who continued mepolizumab treatment in study 205203, in which 94% (47/50) of patients did not experience a flare.
In the 72 patients requiring OCS during Weeks 0 to 4 of the OLE, 28% of patients achieved a mean daily dose OCS dose reduction of ≥50% during Weeks 16 to 20.
Paediatric population: Severe refractory eosinophilic asthma: In MEA115588 and in the double-blind placebo-controlled study 200862, there were 34 adolescents (12 to 17 years old). Of these 34 subjects: 12 received placebo, 9 received mepolizumab 75 mg intravenously, and 13 received 100 mg subcutaneously. In a combined analysis of these studies, a 40% reduction in clinically significant exacerbations was observed in adolescents following mepolizumab treatment compared to placebo (rate ratio 0.60; 95% CI: 0.17, 2.10).
Eosinophilic Granulomatosis with Polyangiitis (EGPA): There are no clinical data available in children and adolescents aged 6 to 17 years old.
HES: Four adolescents (12 to 17 years old) were enrolled in study 200622; one adolescent received mepolizumab 300 mg, and 3 adolescents received placebo for 32 weeks. The one adolescent treated with mepolizumab in the 32-week Study 200622 did not have a HES flare. All 4 adolescents that completed study 200622 continued into a 20-week open-label extension study 205203 in which one of the 4 adolescents experienced one HES flare.
Pharmacokinetics: Following subcutaneous dosing in patients with asthma and CRSwNP, mepolizumab exhibited approximately dose-proportional pharmacokinetics over a dose range of 12.5 mg to 250 mg. Subcutaneous administration of mepolizumab 300 mg had approximately three times the systemic exposure of mepolizumab 100 mg. Following administration of a single 100 mg subcutaneous dose in healthy subjects, mepolizumab systemic exposure was comparable between formulations.
Absorption: Following subcutaneous administration to healthy subjects or patients with asthma, mepolizumab was absorbed slowly with a median time to reach maximum plasma concentration (T
max) ranging from 4 to 8 days.
Following a single subcutaneous administration in the abdomen, thigh or arm of healthy subjects, mepolizumab absolute bioavailability was 64%, 71% and 75%, respectively. In patients with asthma, the absolute bioavailability of mepolizumab administered subcutaneously in the arm ranged from 74-80%. Following repeat subcutaneous administration every 4 weeks, there is approximately a two-fold accumulation at steady state.
Distribution: Following a single intravenous administration to patients with asthma, mepolizumab distributes into a mean volume of distribution of 55 to 85 mL/kg.
Biotransformation: Mepolizumab is a humanized IgG1 monoclonal antibody degraded by proteolytic enzymes which are widely distributed in the body and not restricted to hepatic tissue.
Elimination: Following a single intravenous administration to patients with asthma, the mean systemic clearance (CL) ranged from 1.9 to 3.3 mL/day/kg, with a mean terminal half-life of approximately 20 days. Following subcutaneous administration of mepolizumab, the mean terminal half-life (t1/2) ranged from 16 to 22 days. In the population pharmacokinetic analysis, estimated mepolizumab systemic clearance was 3.1 mL/day/kg.
Special populations: Elderly patients (≥65 years old): There are limited pharmacokinetic data available in elderly patients (≥65 years old) across all clinical studies (N=90). However, in the population pharmacokinetic analysis, there were no indications of an effect of age on the pharmacokinetics of mepolizumab over the age range of 12 to 82 years.
Renal impairment: No formal studies have been conducted to investigate the effect of renal impairment on the pharmacokinetics of mepolizumab. Based on population pharmacokinetic analyses, no dose adjustment is required in patients with creatinine clearance values between 50-80 mL/min. There are limited data available in patients with creatinine clearance values <50 mL/min.
Hepatic impairment: No formal studies have been conducted to investigate the effect of hepatic impairment on the pharmacokinetics of mepolizumab. Since mepolizumab is degraded by widely distributed proteolytic enzymes, not restricted to hepatic tissue, changes in hepatic function are unlikely to have any effect on the elimination of mepolizumab.
Paediatric population: Severe eosinophilic asthma and HES: There are limited pharmacokinetic data available in the paediatric population (59 patients with eosinophilic esophagitis, 55 patients with severe refractory eosinophilic asthma and 1 patient with HES). Intravenous mepolizumab pharmacokinetics was evaluated by population pharmacokinetic analysis in a paediatric study conducted in patients aged 2-17 years old with eosinophilic esophagitis. Paediatric pharmacokinetics was largely predictable from adults, after taking into account bodyweight. Mepolizumab pharmacokinetics in adolescent patients with severe refractory eosinophilic asthma or HES included in the phase 3 studies were consistent with adults (see Dosage & Administration).
Toxicology: Preclinical safety data: As mepolizumab is a monoclonal antibody, no genotoxicity or carcinogenicity studies have been conducted.
Animal toxicology and/or pharmacology: Non-clinical data reveal no special hazards for humans based on conventional studies of safety pharmacology or repeated dose toxicity studies in monkeys. Intravenous and subcutaneous administration to monkeys was associated with reductions in peripheral and lung eosinophil counts, with no toxicological findings.
Eosinophils are thought to be associated with immune system responses to some parasitic infections. Studies conducted in mice treated with anti-IL-5 antibodies or genetically deficient in IL-5 or eosinophils have not shown impaired ability to clear parasitic infections. The relevance of these findings for humans is unknown.
Fertility: No impairment of fertility was observed in a fertility and general reproduction toxicity study in mice performed with an analogous antibody that inhibits IL-5 in mice. This study did not include a littering or functional offspring assessment.
Pregnancy: In monkeys, mepolizumab had no effect on pregnancy or on embryonic/fetal and postnatal development (including immune function) of the offspring. Examinations for internal or skeletal malformations were not performed. Data in cynomolgus monkeys demonstrate that mepolizumab crossed the placenta.
Concentrations of mepolizumab were about 1.2-2.4 times higher in infants than in mothers for several months post partum and did not affect the immune system of the infants.




 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image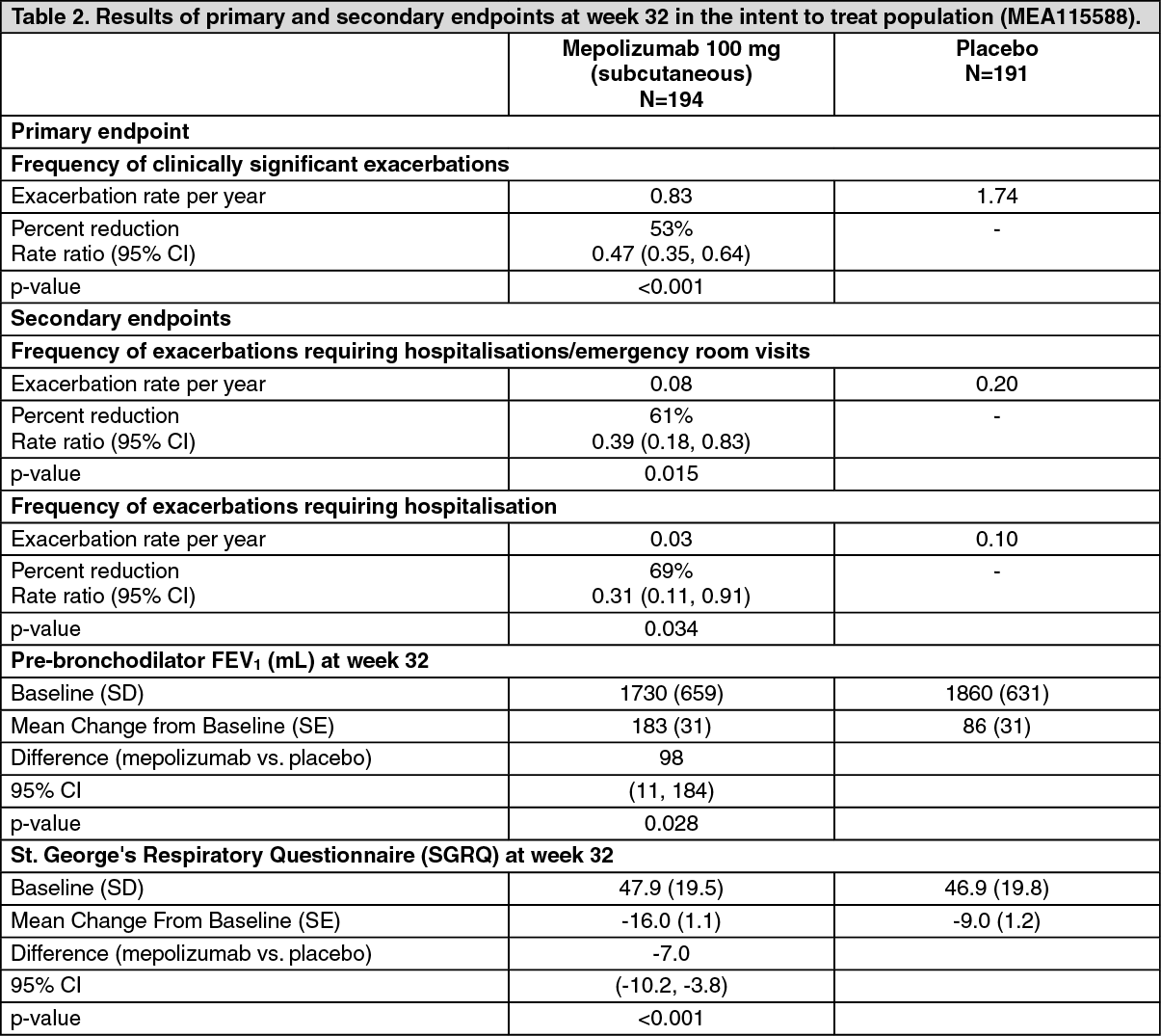 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image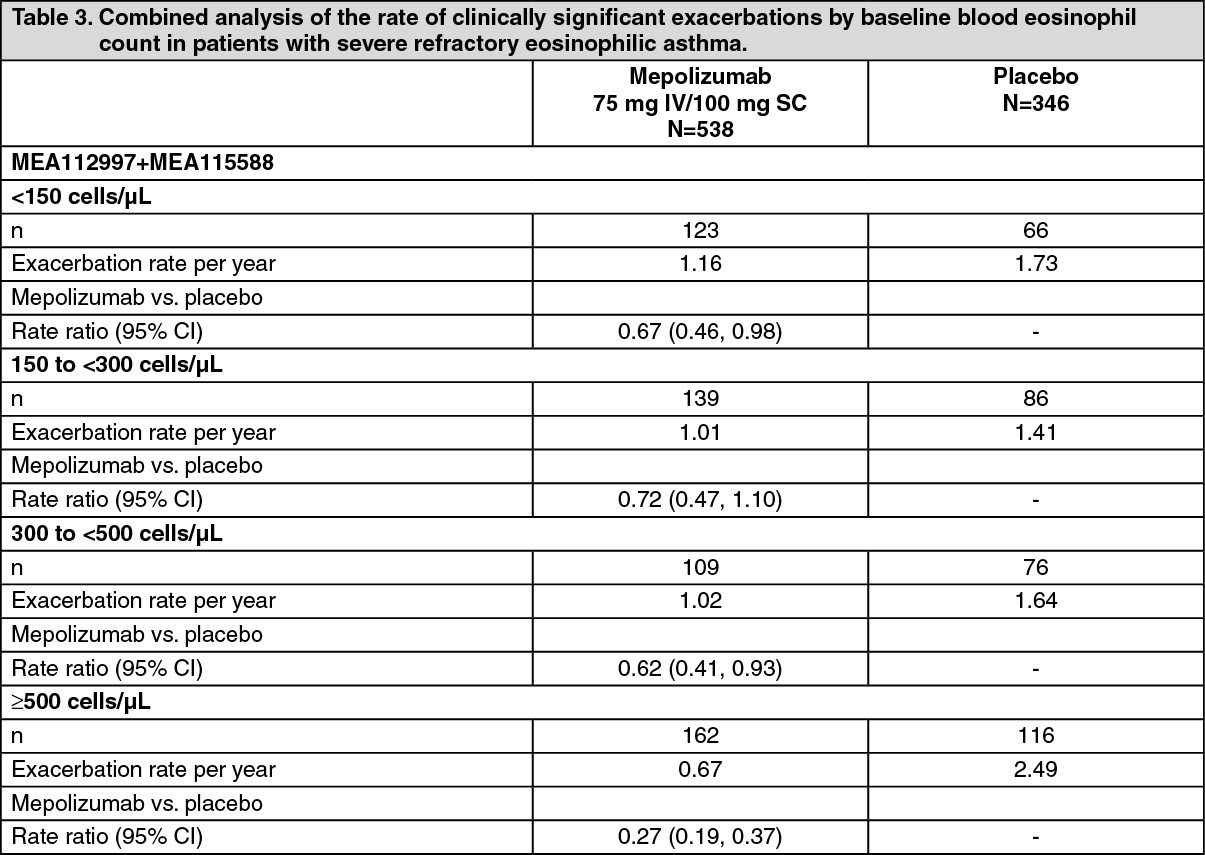 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image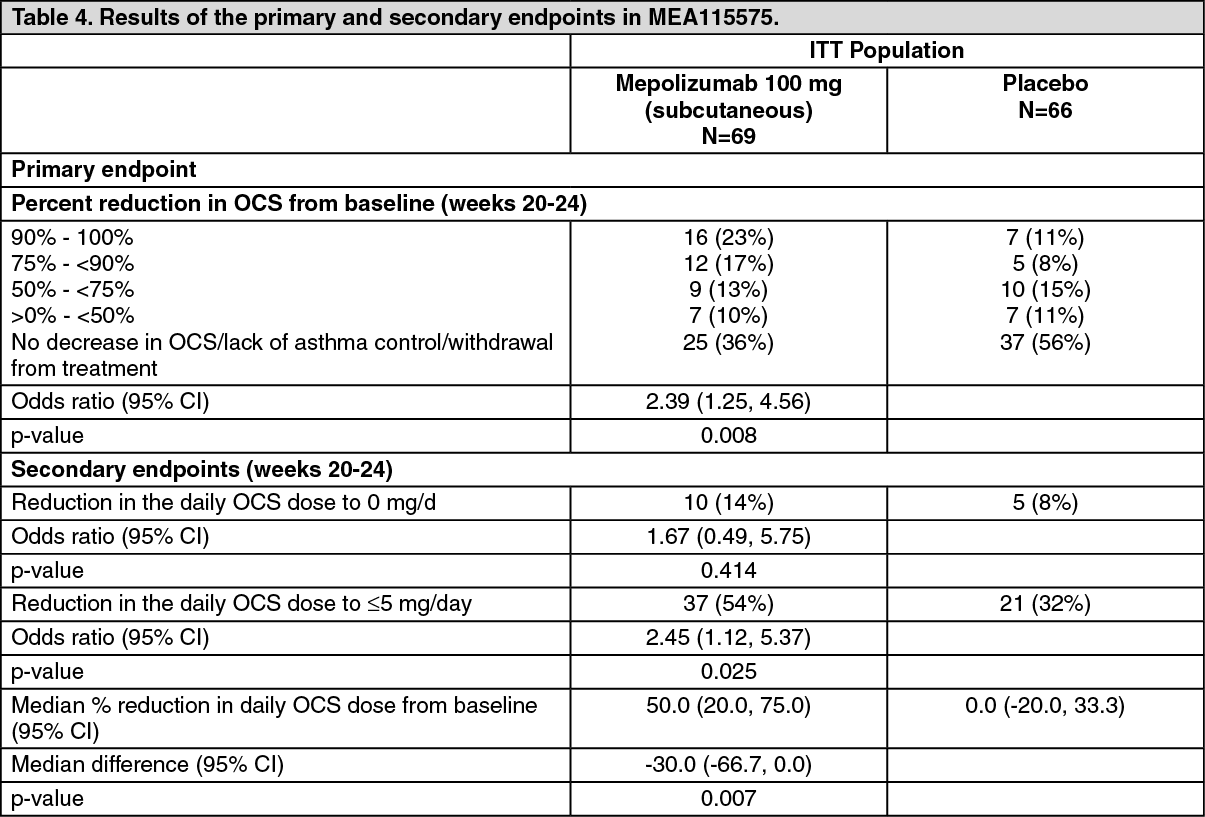 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image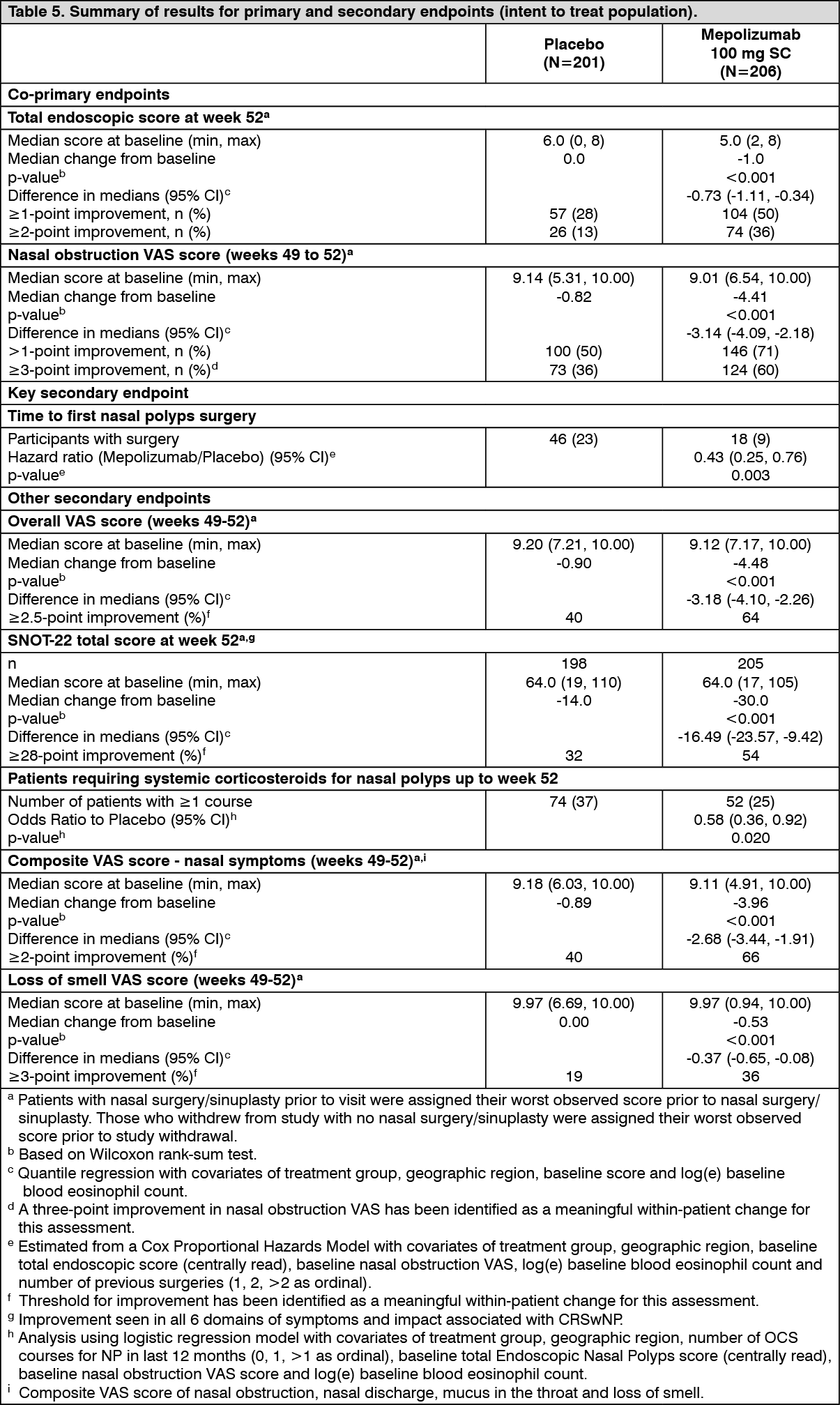 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image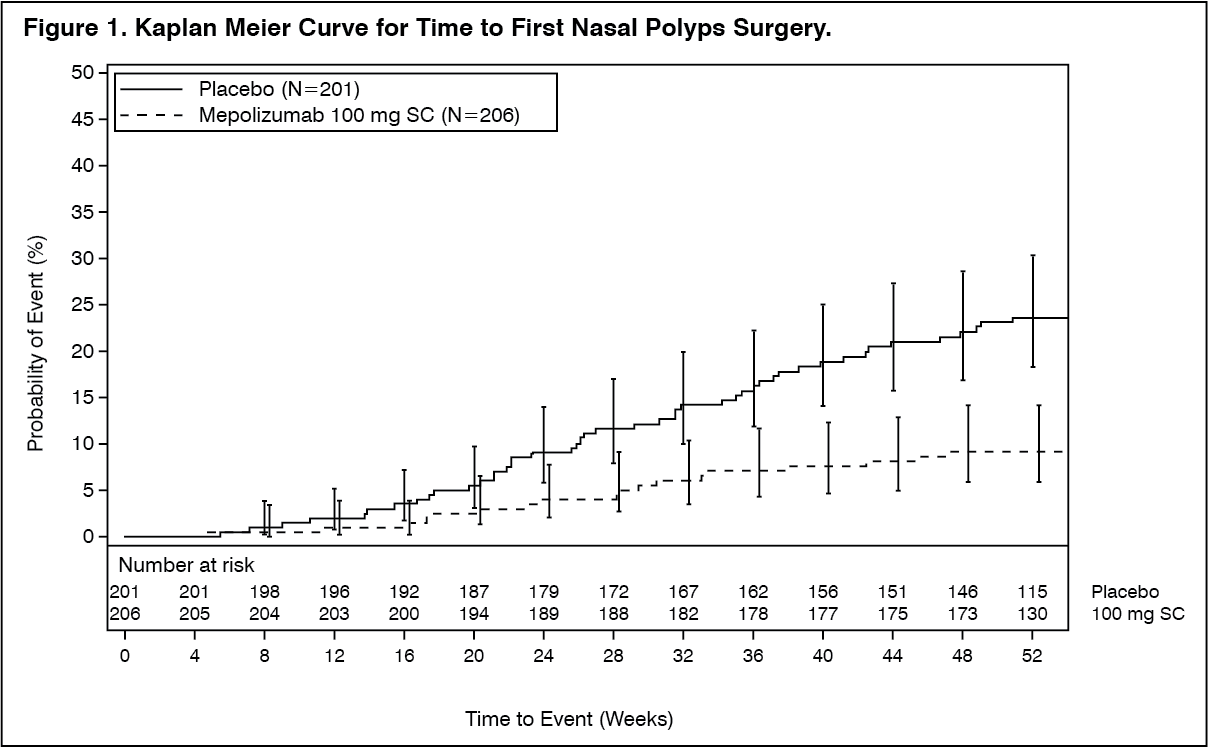 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image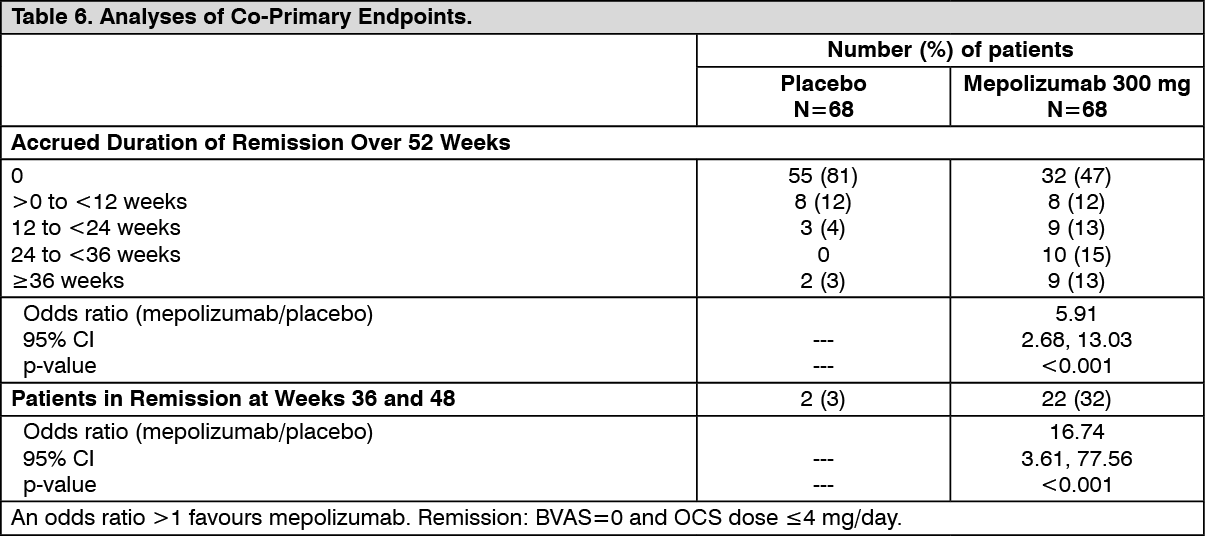 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image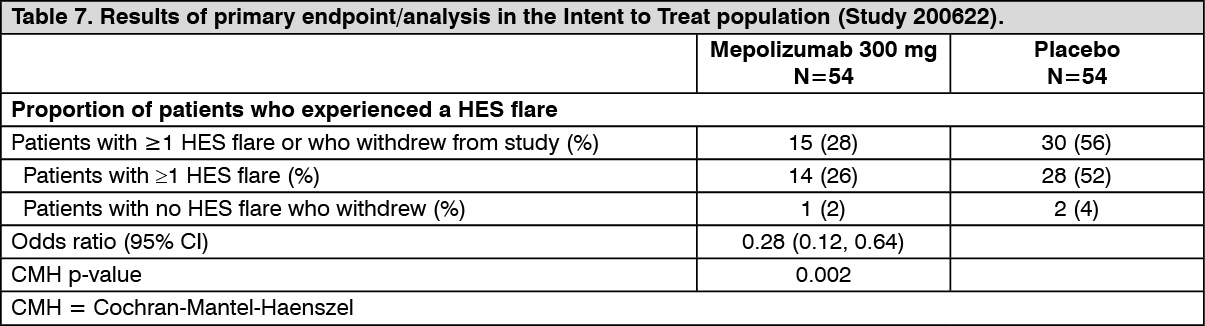 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image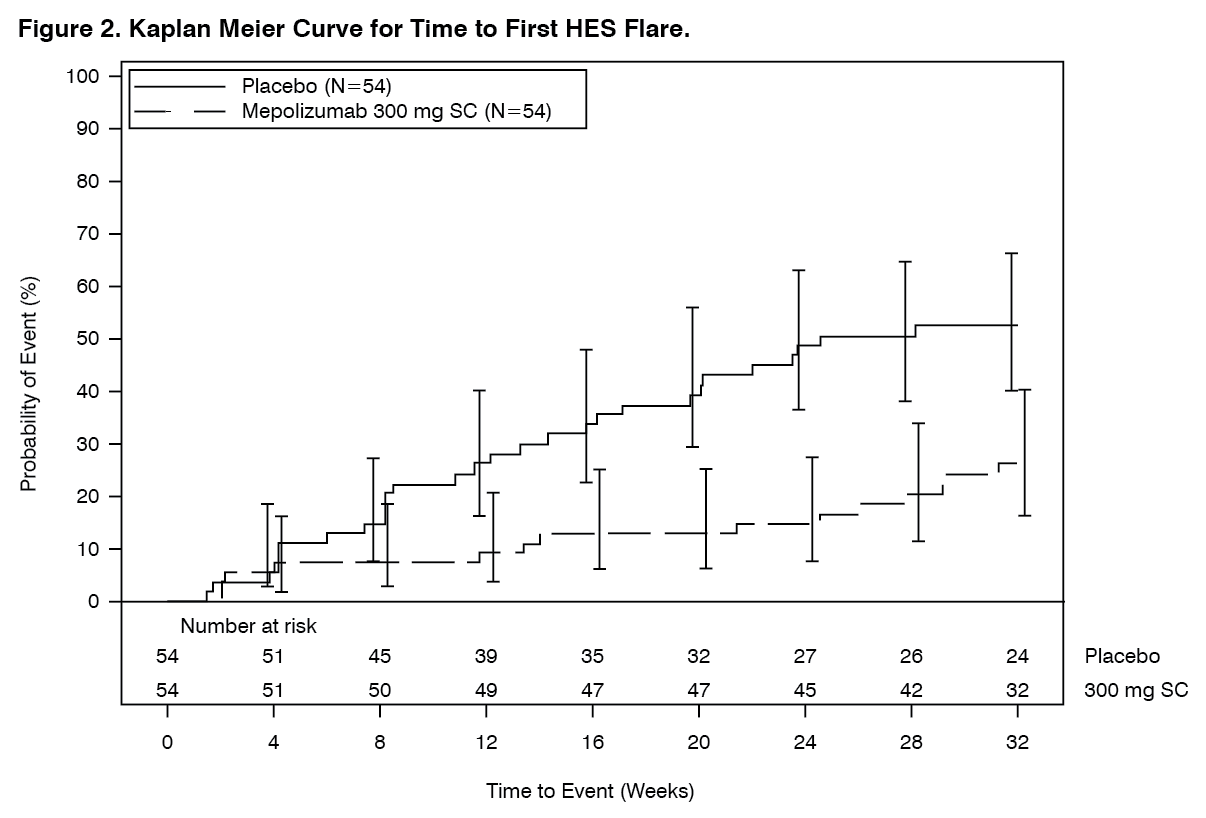 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image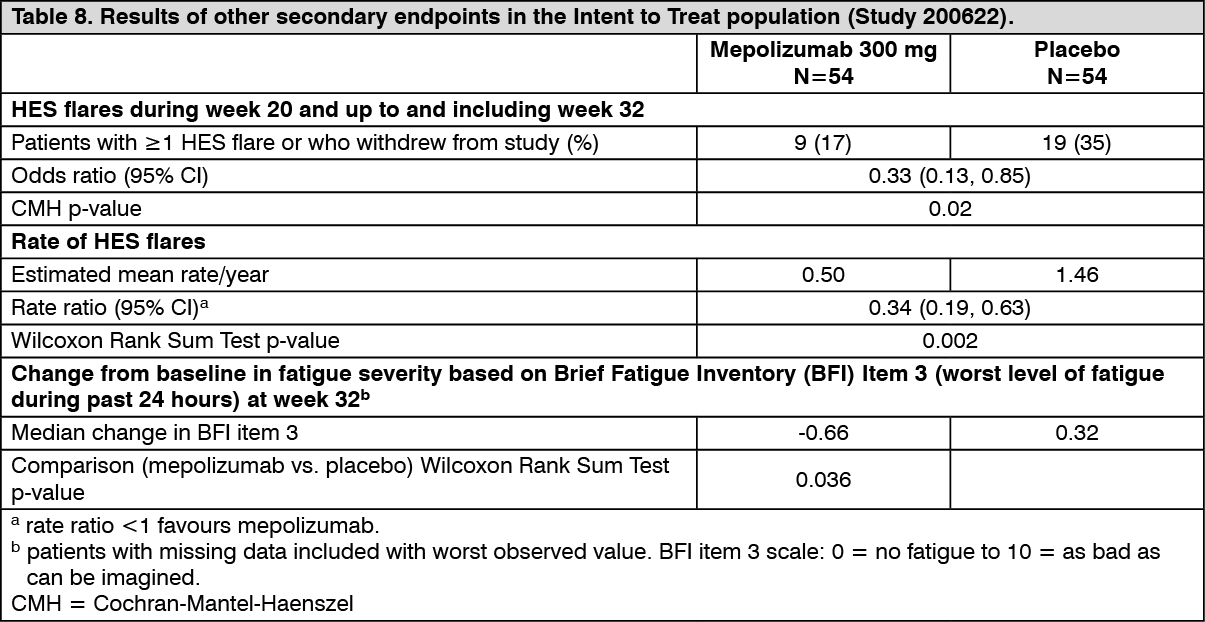 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image


 Sign Out
Sign Out
