


 Sign Out
Sign Out
Mechanism of Action: Citalopram is a highly selective and potent serotonin (5-hydroxytryptamine, 5-HT) reuptake inhibitor with minimal effects on the neuronal reuptake of norepinephrine (NE) and dopamine (DA). The ability of citalopram to potentiate serotonergic activity in the central nervous system via inhibition of the neuronal reuptake of serotonin is thought to be responsible for its antidepressant action. Tolerance to the inhibition of serotonin reuptake is not induced by long-term (14 days) treatment of rats with citalopram.
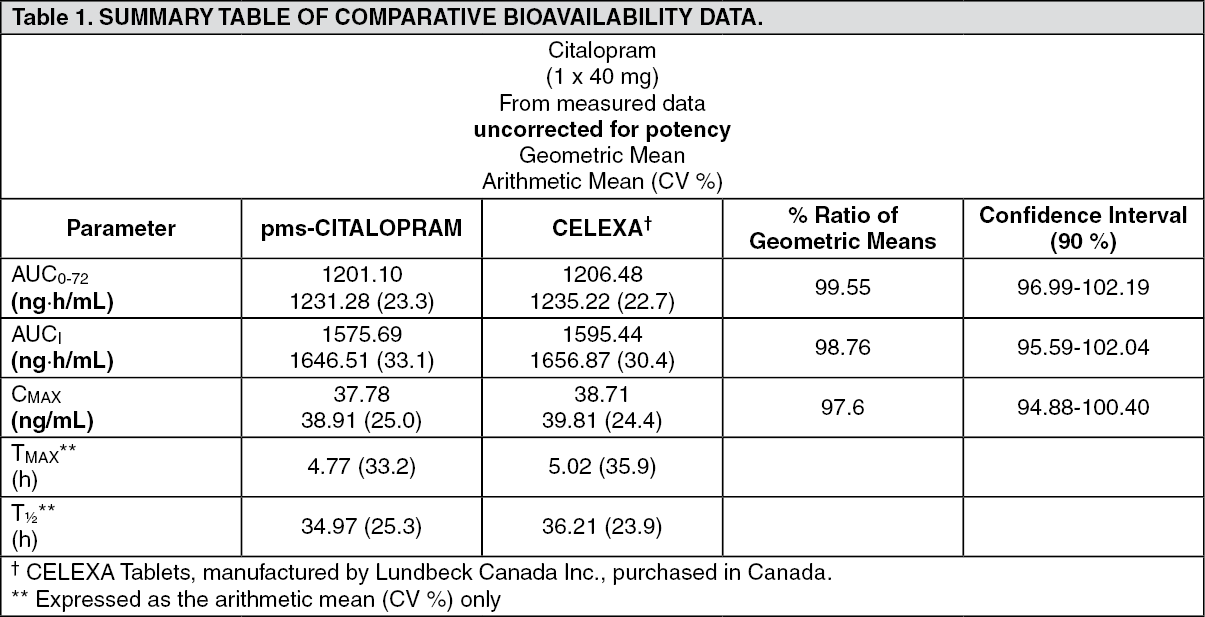
Clinical Trials: Comparative Bioavailability Studies: A blind, randomized, 2-way crossover, bioequivalence study between Pharmascience's pms-CITALOPRAM 40 mg tablets was performed versus Lundbeck's Celexa, administered as 1x 40 mg tablets in 22 healthy male volunteers under fasting conditions. Pharmacokinetic and bioavailability data are presented in the following table: See Table 1.
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageStudy Results: The efficacy of citalopram hydrobromide in the treatment of depression was established in five placebo-controlled studies in patients who met the DSM-III or DSM-III-R criteria for major depression. Response to treatment was evaluated by the Hamilton Depression Rating Scale (HAMD) and/or the Montgomery Asberg Depression Rating Scale (MADRS), as well as the Clinical Global Impression (CGI) Severity Scale. On the HAMD and MADRS, total scores, selected single items, and percentage of responders (defined as patients whose HAMD/MADRS total score decreased by at least 50% versus baseline) were assessed.
In a 6-week fixed-dose, dose-response study, patients received citalopram hydrobromide, at doses of 10, 20, 40, or 60 mg/day or placebo (n=129 to 131 per group). The 40 and 60 mg/day doses were titrated, with patients reaching these designated doses within 4 and 8 days, respectively. The study showed that the 40 and 60 mg/day doses were significantly more effective than placebo, although the 60 mg/day dose was not more effective than the 40 mg/day dose. The lower doses did not show statistically significant superiority over placebo, except on the MADRS; on this scale the percent of 'responders' was significantly higher in all the citalopram hydrobromide-treated groups than in the placebo-treated group.
The second study was a 4-week flexible-dose study in which 85% of the depressed patients met the criteria for melancholia. At entry, 89 and 91 patients were randomized to the citalopram hydrobromide and placebo groups, respectively. This was the only study in which more male than female patients participated (64% vs. 36%). The initial dose of citalopram hydrobromide, 20 mg/day, could be titrated to the maximal tolerated dose or a maximum dose of 80 mg/day. Patients treated with citalopram hydrobromide showed significantly greater improvement than patients treated with placebo. At week 4, the average daily dose was 63 mg, with 52% of patients receiving the 80 mg/day dose.
In a 6-week fixed-dose study, patients received citalopram hydrobromide, 20 or 40 mg/day, or placebo (n=64 to 70 per group). Patients treated with citalopram hydrobromide, 40 mg/day, showed significantly greater improvement than placebo-treated patients. The difference between the lower dose of citalopram hydrobromide and placebo was not significant.
In another 6-week fixed-dose study, patients received citalopram hydrobromide, 20 or 40 mg/day or placebo (n=88 to 97 per group). Although citalopram hydrobromide-treated patients improved to a somewhat greater degree than the placebo-treated patients, the differences between drug and control groups did not reach statistical significance due to a high placebo response, i.e., substantial improvement in the placebo group.
A 6-week, flexible-dose study was conducted in elderly, depressed patients (the mean age of male and female patients was 75 and 77 years, respectively) to determine the antidepressant effect and safety of citalopram hydrobromide in this subpopulation. The number of patients who received citalopram hydrobromide and placebo was 98 and 51, respectively. The study allowed patients to enter with lower baseline HAMD scores than are usually acceptable (≥18 in clinical trials). However, only a small percentage of patients had HAMD scores of less than 18 at entry. The dose of citalopram hydrobromide was titrated from a starting dose of 10 mg/day to a maximum dose of 30 mg/day. Patients treated with citalopram hydrobromide showed significantly greater improvement than patients treated with placebo. The final dose of citalopram hydrobromide was 10, 20 and 30 mg/day in 5%, 51% and 44% of patients, respectively.
The effectiveness of citalopram hydrobromide in preventing relapse was assessed in two long-term studies. Depressed patients who responded to citalopram hydrobromide during an initial 6 or 8 weeks of acute treatment (fixed doses of 20 or 40 mg/day in one study and flexible doses of 20-60 mg/day in the second study) were randomized to continue on citalopram hydrobromide or receive placebo. The number of patients who received citalopram hydrobromide and placebo was 257 and 116, respectively. In both studies, patients who continued on citalopram hydrobromide experienced significantly lower relapse rates over the subsequent 6 months compared to those receiving placebo. In the fixed-dose study, the relapse rates were similar at the 20 and 40 mg/day doses, namely 10% and 12%, respectively. Of the placebo-treated patients, 31% experienced relapse. In the flexible-dose study, the relapse rates were 14% and 24% in the citalopram hydrobromide- and placebo-treated patients, respectively. While the majority of patients (76%) were maintained on 20 or 40 mg/day of citalopram hydrobromide during most of the study, some patients received 60 mg/day, while a few patients were maintained on less than 20 mg/day.
Detailed Pharmacology: Pharmacodynamics: Citalopram is a racemic mixture with the S (+) enantiomer mediating the pharmacological effects. The R (-) enantiomer contributes little to the activity of citalopram.
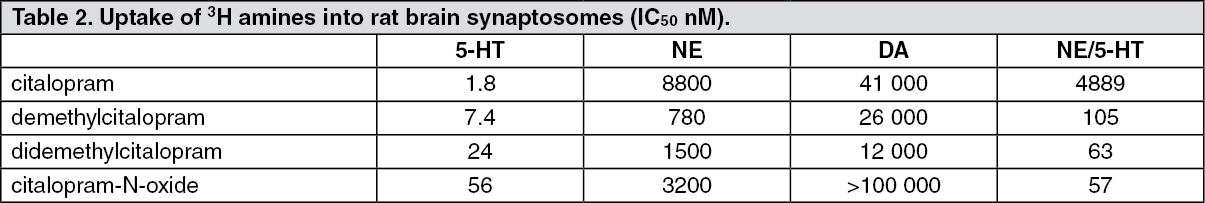
In Vitro Experiments: (a) Neuronal reuptake of serotonin, norepinephrine and dopamine: The primary pharmacological effect of citalopram is inhibition of the 5-HT reuptake mechanism. Citalopram was shown to inhibit 5-HT uptake in rabbit blood platelets, with an IC50 of 14 nM. Similarly, the drug inhibits 5-HT uptake in rat brain synaptosomal preparations. (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe data indicate that citalopram is a potent and specific 5-HT uptake inhibitor with no activity on the neuronal reuptake of norepinephrine (NE) or dopamine (DA). The metabolites of citalopram are also specific inhibitors of 5-HT reuptake, albeit less active than the parent drug.
The ratio between the concentrations inhibiting the in vitro uptake of NE and 5-HT determine the selectivity of a SSRI. According to this criterion citalopram is a highly selective SSRI.
(b) Effect on neurotransmitter receptors: Citalopram has no or very low affinity for a series of receptors including 5-HT1A, 5-HT2, dopamine D1 and D2 receptors, α1-, α2-, β-adrenoreceptors, histamine H1, muscarinic cholinergic, benzodiazepine, and opioid receptors.
A series of functional in vitro tests in isolated organs as well as functional in vivo tests have confirmed the lack of receptor affinity.
Behavioral Effects: In a "behavioral despair paradigm", mice, trained to swim in a glass jar, eventually exhibit immobility. This behavior was dose-dependently reversed by citalopram.
The 5-HT precursors, tryptophan and 5-HTP, induce in mice and rats the 5-HT syndrome, characterized by tremor, hyperactivity, abnormal gait, lordosis, and abduction of the hind limbs. Citalopram potentiated these behavioral manifestations. The demethyl, didemethyl, and N-oxide metabolites were less potent than the parent drug.
The characteristic head twitches, induced by a combined treatment with a MAOI and 5-HTP, were potentiated by citalopram. However, head twitches induced by quipazine, a direct 5-HT mimetic, were not affected by citalopram, indicating that the drug has no anti-5-HT activity.
Although citalopram has no antinociceptive activity per se, it potentiated the antinociceptive effect of morphine. In a food reinforcement paradigm, delivered under a multiple schedule, citalopram did not affect the responding in pigeons but potentiated the 5-HTP-induced decrease in responding.
In rats, citalopram did not facilitate self-stimulation, did not substitute for d-amphetamine, d-LSD, or 8-OHDPAT in a drug discrimination paradigm and did not increase ethanol consumption in an ethanol/water preference test. In the latter experiment, citalopram actually decreased ethanol consumption. These experiments indicate that citalopram would not be abused and would not cause dependence.
Citalopram had a slight protective effect against maximal electroshock-induced convulsions, isoniazide-induced convulsions and audiogenic seizure. However, in toxicity studies convulsions have been observed at very high plasma levels of citalopram (see Toxicology as follows).
Cardiovascular Effects: Citalopram blocked heterologous HERG-mediated currents in transfected Chinese hamster ovary cells with an IC50 of 4 mcM.
In conscious dogs, single oral doses of 5 mg/kg of citalopram caused pronounced fluctuation of the blood pressure and heart rate. A 10 mg/kg dose caused tachycardia and elevated blood pressure. The ECG was unchanged.
In anaesthetized cats, single oral doses of 35 mg/kg decreased the following parameters: mean arterial blood pressure, left ventricular end diastolic pressure, contractility, cardiac performance, stroke volume, and cardiac output. Peripheral resistance was increased. ECG abnormalities included alterations in conduction, changes in rhythm and T-wave inversion in 2 of 6 cats.
Additional cardiovascular effects of citalopram and a metabolite are described under Toxicology.
Pharmacokinetics: Absorption: Following the administration of a single oral dose of citalopram (40 mg) to healthy male volunteers, peak blood levels occurred at about 4 hours (range 1 to 6 hours). The absolute bioavailability of citalopram was about 80% (range 52 to 93%) relative to an intravenous dose. Absorption was not affected by food.
Distribution: After intravenous infusion in healthy male volunteers, the apparent volume of distribution (Vd) β was about 12 L/kg (range 9-17 L/kg), indicating a pronounced tissue distribution; (Vd)β oral was about 17 L/kg (range 14-21 L/kg). The binding of citalopram and its demethylated metabolites to human plasma proteins is about 80%.
The single- and multiple-dose pharmacokinetics of citalopram are linear and dose-proportional in a dose range of 10 to 60 mg/day. Steady-state plasma levels are achieved in patients in 1-2 weeks. At a daily dose of 40 mg, the average plasma concentration is about 83 ng/mL (n=114) with a range from 30 to 200 ng/mL. Citalopram does not accumulate during long-term treatment. A clear relationship between citalopram plasma levels and therapeutic response or side effects has not been established.
Metabolism: Citalopram is metabolized in the liver to demethylcitalopram (DCT), didemethylcitalopram (DDCT), citalopram-N-oxide, and a deaminated propionic acid derivative.
In vitro studies show that DCT, DDCT and citalopram-N-oxide also inhibit the neuronal reuptake of serotonin but are less selective and less potent than the parent compound and are of minor clinical importance. Unchanged citalopram is the predominant compound in plasma.
In vitro studies indicated that the biotransformation of citalopram to its demethyl metabolites depends on both CYP2C19 and CYP3A4, with a small contribution from CYP2D6. An initial dose of 10 mg is recommended for known poor metabolisers of CYP2C19 (see Recommended Dose and Dosage Adjustment: CYP2C19 Poor Metabolisers under Dosage & Administration).
Elimination: The elimination half-life of citalopram (t2β) is approximately 37 hours (range: 30 - 42 hours) which allows recommendation of once-daily dosing. The systemic citalopram plasma clearance (ClS) is 0.33 L/min. Citalopram is eliminated primarily via the liver (85%) and the remainder via the kidneys; approximately 12% (range 6-21%) of the daily dose is excreted in urine as unchanged citalopram.
Special Populations and Conditions: Geriatrics: Elderly patients (4 males and 7 females aged 73 - 90 years), received a 20 mg/day dose of citalopram for 3-4 weeks. In the elderly, steady state plasma levels were elevated (106 ng/mL), half-life prolonged (1.5 - 3.75 days) and clearance decreased (0.08 - 0.3 L/min). Elevation of citalopram plasma levels occurred at an earlier age in females than in males. In this population, lower doses and a lower maximum dose of citalopram are recommended (see Recommended Dose and Dosage Adjustment: Geriatrics (≥ 65 years of age) under Dosage & Administration and Use in Elderly under Precautions).
Hepatic Insufficiency: The pharmacokinetics of citalopram were compared in patients with reduced hepatic function (3 female and 6 male patients aged 41 - 60 years) to those seen in 12 healthy male volunteers (aged 21 - 43 years). In patients with reduced hepatic function the half-life of citalopram was approximately doubled (83 hours vs. 37 hours), steady state citalopram concentrations increased by 61% and oral clearance decreased by 37%. Consequently, the use of citalopram in patients with reduced hepatic function should be approached with caution and lower maximal doses should be prescribed (see Recommended Dose and Dosage Adjustment: Hepatic Impairment under Dosage & Administration and Hepatic/Biliary/Pancreatic: Hepatic Impairment under Precautions).
Renal Insufficiency: In patients with mild to moderate reduction of the renal function (4 female and 3 male patients aged 30-55 years), citalopram was being eliminated more slowly than in 12 healthy male volunteers (aged 21-43 years); half-lives being 49 hours vs. 37 hours. However, mild to moderate renal impairment had no major influence on the kinetics of citalopram. At present, no information is available for chronic treatment of patients with severely reduced renal function (creatinine clearance <20 mL/min).
Pharmacokinetics in Animals: Absorption: The kinetics of citalopram in mouse, rat, and dog are characterized by rapid absorption, with Tmax ranging from 0.5 to 4 hours. In contrast to man, reduced systemic bioavailability due to extensive first-pass metabolism has been demonstrated in animals.
Distribution: Pharmacokinetic analysis of single dose i.v. data suggests two-compartment distribution characteristics. High levels of drug and demethylated metabolites were found in the lungs, liver, and kidneys, and lower levels in the heart and brain. Citalopram and the demethylated metabolites were shown to pass the placental barrier and were excreted in small amounts in milk.
The plasma protein binding of citalopram has been estimated to be 70-80%. The binding protein(s) has not been identified.
Both in mice and dogs, tissue concentrations of parent drug as well as those of the demethylated metabolites increased with increasing doses, although not necessarily in a dose-related manner. Levels of the didemethylated metabolites were higher in dogs than in mice in relation to the parent drug, resulting in smaller citalopram/didemethylcitalopram ratios in the dog, particularly in the heart and kidneys.
Metabolism: There are no major qualitative differences in the metabolism of citalopram between animals and man. Citalopram is metabolized to demethylcitalopram, didemethylcitalopram, citalopram-N-oxide, and the deaminated propionic acid.
Demethylcitalopram and didemethylcitalopram levels are more prominent in mouse, rat, and dog than in man.
Elimination: Elimination of citalopram after a single dose is rapid, the half-life ranging from 1.5-2 hours in the mouse to 3.5-8 hours in the dog. In the dog, the half-life is prolonged with increasing doses due to saturation of the first-pass metabolism.
Following the administration of 14C-labelled citalopram to rats, at a dose of 20 mg/kg, approximately equal amounts of the dose were excreted in the urine and feces, with total recovery being about 80%.
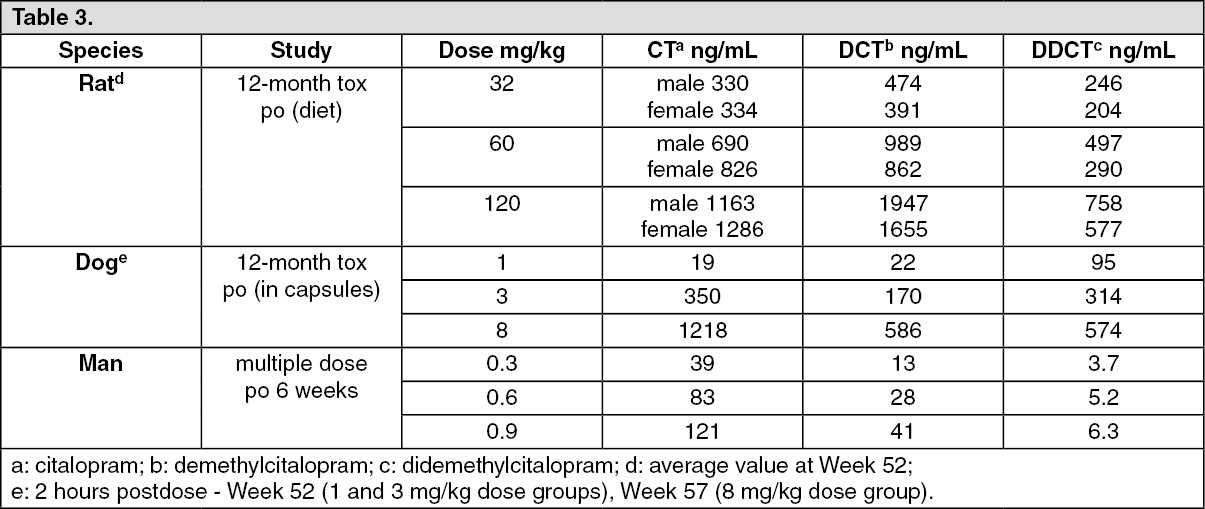
Toxicokinetics: Plasma levels were determined in several long-term toxicity studies. The following table summarizes the results seen in some of these studies. (See Table 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe data indicate that the plasma levels of citalopram, as well as those of the demethylated metabolites, are considerably higher in animals than in man. The approximate 0.9 mg/kg dose in man corresponds to the highest recommended dose (60 mg/day). The plasma levels of the parent drug, seen in rats and dogs at the highest doses, are approximately 10 times higher in animals than in man, while the levels of the didemethyl metabolites are almost 100 fold higher. In the rat, a NOEL (no observable effect level) could not be established in this study; at the low dose minimal vacuolization of hepatocytes with fatty infiltration, and foam cell accumulation in lungs were noted. The changes were reversible. In dogs, the NOEL was 3 mg/kg.
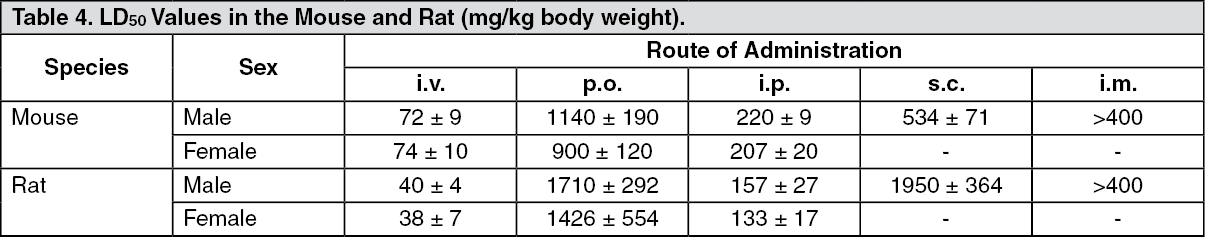
Toxicology: Acute Toxicity: The LD50 values of citalopram ranged between 900-1700 mg/kg after oral administration and 38-74 mg/kg after intravenous administration. However, some mortality was also seen in the 400-600 mg/kg dose range, indicating a very flat dose-response curve regarding mortality. Signs of toxicity were sedation and tremor, while convulsions occurred at doses close to or above the LD50 values. (See Table 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageA number of single dose toxicity studies have been carried out in dogs to investigate the potential cardiovascular toxicity of citalopram. In these studies, cardiotoxicity was not observed, but tonic-clonic convulsions were seen after oral administration of 20-40 mg/kg, as well as after slow intravenous infusion of 20-24 mg/kg. The critical plasma concentration for convulsions was about 1950 ng/mL.
Long-term Toxicity: Toxicological studies, including daily dosing for periods up to 26 weeks in mice and 52 weeks in rats and dogs, have been carried out. Plasma drug monitoring in the long-term safety studies documented that animals have been exposed to average citalopram levels of up to about 1200 ng/mL (dogs and rats) and 2900 ng/mL (mice), as well as substantial levels of demethylcitalopram [up to about 1800 ng/mL (rats), 600 ng/mL (dogs), 1150 ng/mL (mice)] and didemethylcitalopram [up to about 650 ng/mL (rats), 600 ng/mL (dogs), 300 ng/mL (mice)].
Apart from behavioral and functional characteristics of exaggerated 5-HT stimulation (e.g., hyperactivity, tremor, tail rigidity, mydriasis, reduced food consumption, and reduced body weight gain), two treatment-related findings have been demonstrated in rodents, namely fatty infiltration of the liver and lipidosis (vacuolization of lymphocytes). Both of the findings were reversible. In addition, retinal degeneration and testicular atrophy were also observed in rats.
In dogs, two treatment-related effects were found. Firstly, convulsions and death when plasma citalopram levels exceeded 1950 ng/mL (p.o. or i.v.). Secondly, fatal ventricular arrhythmias at combined high levels of the didemethyl metabolite (about 300 ng/mL) and citalopram (about 1950 ng/mL) were seen following i.v. infusion.
Hepatic Fatty Infiltration in Rodents: Fatty infiltration in the liver was first observed in a 3-month gavage study in rats given 8-32 mg/kg/day of citalopram. This administration resulted in dose-related hepatic fatty infiltration in all male rats but not in female rats at any of the doses. The fatty infiltration in male rats was also observed in a 4-week study, however, only at considerably higher doses (>160 mg/kg). In female rats only minimal fatty infiltration was seen at a 200 mg/kg/day dose.
Lipidosis (phospholipids) in Rodents: Phospholipidosis, which has been seen in rodents, is an abnormal accumulation of phospholipids in phagocytic cells and cells which catabolize biomembranes, such as pulmonary alveolar macrophages and circulating leucocytes (especially lymphocytes).
Phospholipidosis developed in rats receiving citalopram at daily doses of 120 mg/kg and slight vacuolization of peripheral lymphocytes was observed in mice at daily doses of 100 mg/kg, in the 52-week and 26-week studies, respectively. Both conditions were reversible within 3-4 weeks.
Retinal Degeneration/Atrophy in Rats: In the rat carcinogenicity study, a slight, dose-related increase in lens opacity was seen, affecting males only. In addition, increased incidence/severity of retinal degeneration/atrophy was seen in the high-dose group (80 mg/kg/day). The incidence was higher in females, however, more female than male rats survived the study. It was concluded by an independent pathologist that the retinal changes were most likely related to drug-induced pupillary dilatation (mydriasis) which increased the risk of retinal damage in the already light-sensitive albino rat.
Testicular Atrophy in Rats: In the 52-week rat toxicity study, testicular atrophy was seen at the 60 and 120 mg/kg/day doses of citalopram.
Convulsions and Death in Dogs: Toxicity studies in dogs revealed that citalopram administration led to fatal ventricular arrhythmias. Consequently, studies were undertaken to elucidate the mechanism of this effect and to determine its relevance to humans.
The studies have shown that: i.v. infusion of citalopram, at a dose of 20 mg/kg, led to convulsions. The blood levels of citalopram were 1950 ng/mL at this dose. In the presence of diazepam, also infused intravenously, higher doses of citalopram could be infused, namely up to 70 mg/kg (6800 ng/mL).
Intravenous infusion of the didemethyl metabolite of citalopram caused QT prolongation in a dose range of 5 to 22 mg/kg. The blood levels of the metabolite were 300 ng/mL at the 5 mg/kg dose. The QT prolongation was dose-dependent.
When citalopram, 20 mg/kg, and didemethylcitalopram, 5 mg/kg, were infused concomitantly (in the presence of diazepam in order to prevent convulsions), 5 out of 9 dogs died due to ventricular fibrillation. At these doses, the plasma levels of citalopram and didemethylcitalopram were 1950 ng/mL and 300 ng/mL, respectively.
As shown in the following table, there is a substantial difference in the plasma levels of citalopram and its metabolite in dogs and in humans at the recommended therapeutic doses. (See Table 5.)
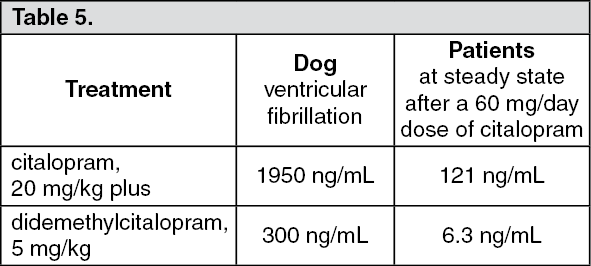 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageReproduction Toxicity: Citalopram did not affect the reproductive performance of rats at dosages up to 16 mg/kg/day (males) and 32 mg/kg/day (females).
In the teratology studies in rats, effects were observed in the conceptuses at dosages that were toxic to the dams. Minimal developmental toxicity was evident at 32 mg/kg/day: manifested as low incidences of resorptions, slightly reduced fetal and pup weights, and small reversible delays in ossification and postnatal development.
In rabbits, dosages of 4.8 mg/kg/day and above were toxic to the dams, and 16 mg/kg/day and above caused deaths. There were no effects on embryo-fetal development at the highest dose that could be assessed (16 mg/kg/day).
In a rat embryo/fetal development study, oral administration of citalopram (32, 56, or 112 mg/kg/day) to pregnant animals during the period of organogenesis resulted in decreased embryo/fetal growth and survival and an increased incidence of fetal abnormalities (including cardiovascular and skeletal defects) at the high dose, which is approximately 18 times the MRHD of 60 mg/day on a body surface area (mg/m2) basis. This dose was also associated with maternal toxicity (clinical signs, decreased body weight gain). The developmental, no-effect dose of 56 mg/kg/day is approximately 9 times the MRHD on a mg/m2 basis. In a second embryo/fetal developmental study in rats conducted at similar dose levels, no increase in fetal abnormalities were observed.
In a rabbit study, no adverse effects on embryo/fetal development were observed at doses of up to 16 mg/kg/day, or approximately 5 times the MRHD on a mg/m2 basis. Thus, teratogenic effects were observed at a maternally toxic dose in one embryo-foetal developmental study in the rats, but were not confirmed in a second rat study or in the rabbit.
When female rats were treated with citalopram (4.8, 12.8, or 32 mg/kg/day) from late gestation through weaning, increased offspring mortality during the first 4 days after birth and persistent offspring growth retardation were observed at the highest dose, which is approximately 5 times the MRHD on an mg/m2 basis. The no-effect dose of 12.8 mg/kg/day is approximately 2 times the MRHD on an mg/m2 basis. Similar effects on offspring mortality and growth were seen when dams were treated throughout gestation and early lactation at doses ≥ 24 mg/kg/day, approximately 4 times the MRHD on an mg/m2 basis. A no-effect dose was not determined in that study.
Fertility: Animal data have shown that citalopram induces a reduction of fertility index and pregnancy index, reduction in number in implantation and abnormal sperm at exposure well in excess of human exposure.
Mutagenic Potential: Citalopram did not have mutagenic activity in most of the in vitro tests (Ames Salmonella assay; chromosome aberration assay in cultured human lymphocytes; gene mutation assay in cultured mouse lymphoma L5178Y) and in vivo tests (micronucleus test; unscheduled DNA synthesis). However, citalopram was mutagenic in some in vitro studies (Ames Salmonella assay and Chinese hamster lung cell assay).
Carcinogenicity: Citalopram did not show any carcinogenic potential in mice at daily doses of 40-240 mg/kg (1.5 years) and in rats at 8-80 mg/kg (2 years). There was an increased incidence of small intestine carcinoma in rats treated with 8 and 24 mg/kg/day of citalopram but not in rats treated with an 80 mg/kg/day dose.