


 Sign Out
Sign Out

Electrocardiography: A double-blind, placebo-controlled, randomised, multiple dose study was performed in two cohorts of healthy adult subjects (CYP2D6 intermediate and extensive metabolizers). In the first cohort, subjects received once-daily oral dosing for 28 days with fluoxetine 20 mg (N = 12) or placebo (N = 4), whilst in the second cohort, subjects received once-daily oral dosing for 28 days with fluoxetine 40 mg (N = 12) or placebo (N = 4). Serial ECG assessments were performed at baseline and on days 1 and 28 of treatment. For the 40 mg fluoxetine treatment (N = 12), the maximal mean difference from placebo in change from time-averaged baseline in QTcF (QT/RR0.33) was 12.005 msec (90% CI 4.412, 19.598) on day 28. For the 20 mg treatment, the corresponding placebo-adjusted increase in the QTcF interval was 4.841 msec (90% CI -4.009, 13.69) (see Cardiovascular under Precautions; Clinical Trial Adverse Drug Reactions: Adults: ECG Findings under Adverse Reactions; Post-Market Adverse Drug Reactions under Adverse Reactions; Drug-Drug Interactions: QTc-Prolonging Drugs under Interactions).
Clinical Trials: Comparative Bioavailability Studies: A comparative bioavailability study of fluoxetine capsules 20 mg was performed. Pharmacokinetic and bioavailability data were measured in 90 volunteers in the fasting state. The results can be summarized as follows: See Table 2.
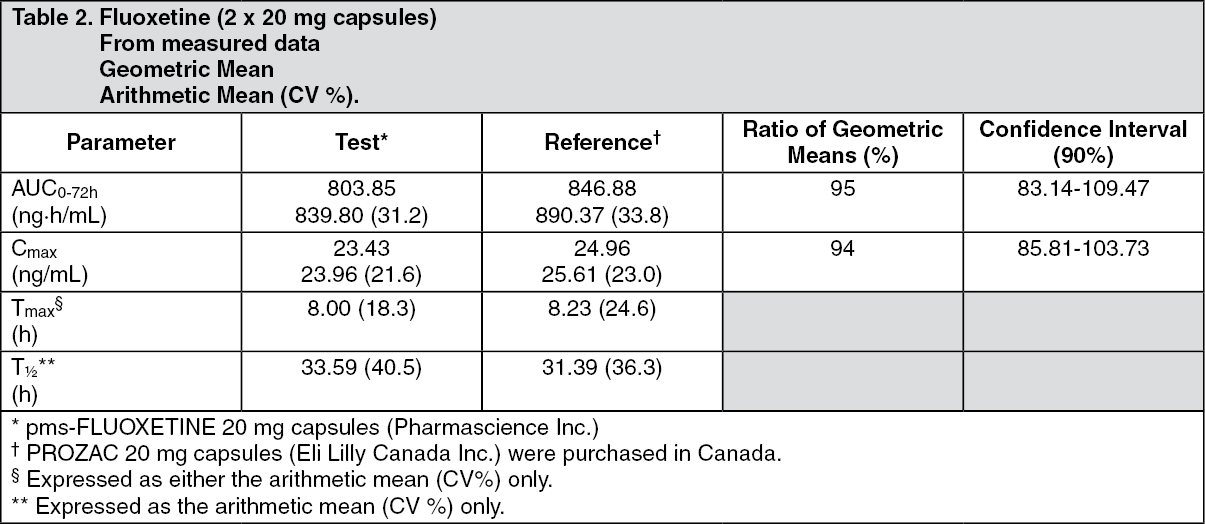 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe efficacy of fluoxetine was established in 5- and 6-week placebo-controlled clinical trials in depressed outpatients (≥18 yr of age), who meet the DSM-III-R criteria for major depressive disorder.
Two, 6-week placebo-controlled clinical trials in depressed elderly patients, who met the DSM-III-R criteria for major depressive disorder (mean age 67.4 yr, range 60 to 85 yr) have shown fluoxetine, 20 mg/day, to be effective.
A study was conducted involving depressed outpatients who had responded (modified HAMD-17 score of ≤7 during each of the last 3 weeks of open-label treatment and absence of major depression by DSM-III-R criteria) by the end of an initial 12-week open treatment phase on fluoxetine 20 mg/day. These patients (N=298) were randomized to continuation on double-blind fluoxetine 20 mg/day or placebo. At 38 weeks (50 weeks total), a statistically significantly lower relapse rate (defined as symptoms sufficient to meet a diagnosis of major depression for 2 weeks or a modified HAMD-17 score of ≥14 for 3 weeks) was observed for patients taking fluoxetine compared to those on placebo.
Detailed Pharmacology: In vitro and in vivo studies have shown fluoxetine and norfluoxetine (the major metabolite) to be potent and selective inhibitors of neuronal pre-synaptic reuptake of serotonin. Serotonin released into the synaptic cleft by a nerve impulse is inactivated principally by reuptake into the presynaptic nerve ending where it is metabolized or retained in storage granules. Fluoxetine specifically inhibits the reuptake process, thereby allowing serotonin to remain longer in the synaptic cleft and enhancing the action of the neurotransmitter on synaptic receptors. Fluoxetine has only weak affinity for various receptor systems in receptor binding studies.
A number of behavioural, neuroendocrinologic, and other pharmacologic effects of fluoxetine in experimental animals have been attributed to its enhancement of serotonergic function by inhibition of serotonin uptake. Fluoxetine restored the capacity for acquisition of passive avoidance task in olfactory bulbectomized rats, potentiated 5-hydroxy-tryptophan-induced head twitch in mice, potentiated 5-hydroxytryptophan-induced depression of operant behaviour in pigeons, and potentiated the behavioural effect of 5-hydroxytryptophan in rats working on a milk reinforcement schedule. Fluoxetine suppressed REM sleep in rats and cats, and reduced the amount or altered the composition of dietary intake in rats. It also selectively reduced non-protein caloric intake in rats.
Few pharmacologic actions of fluoxetine other than inhibition of serotonin uptake and consequences of that inhibition have been found. For instance, fluoxetine does not antagonize reserpine- or apomorphine-induced hypothermia in mice, and does not reduce immobility in the forced swimming test in rats.
Pharmacokinetics: Absorption, Distribution, Metabolism, and Excretion: Fluoxetine is well absorbed after oral administration. In man, following a single oral 40 mg dose, peak plasma concentrations of fluoxetine from 15 to 55 ng/mL are observed after 6 to 8 hours. Food does not appear to affect the systemic bioavailability of fluoxetine, although it may delay its absorption inconsequentially. Thus, pms-FLUOXETINE may be administered with or without food.
Fluoxetine is extensively metabolized in the liver to norfluoxetine, and other unidentified metabolites. The pharmacological activity of norfluoxetine, which is formed by demethylation of fluoxetine appears to be similar to that of the parent drug. Norfluoxetine contributes to the long duration of action of fluoxetine. The primary route of elimination appears to be hepatic metabolism to inactive metabolites excreted by the kidney. The elimination half-life of fluoxetine is 4 to 6 days and that of its active metabolite is 4 to 16 days.
Clinical Issues Related to Metabolism/Elimination: Variability in Metabolism: The metabolism of fluoxetine, like that of a number of other compounds, including tricyclic antidepressants and some selective serotonin reuptake inhibitors (SSRIs), involves the P4502D6 system. Concomitant therapy with fluoxetine and the aforementioned drugs may lead to clinically significant drug interactions (see Drug-Drug Interactions: Drugs Metabolized by P4502D6 Isoenzyme and Impact of CYP2D6 Inhibition on Fluoxetine Metabolism under Interactions).
Accumulation and Slow Elimination: The relatively slow elimination of fluoxetine and its active metabolite, norfluoxetine, results in significant accumulation of these active moieties in chronic use. Therefore, it may take up to 1 to 2 months for the active drug substance(s) to disappear from the body. This persistence of active moieties is important to keep in mind when pms-FLUOXETINE is discontinued, or when drugs that are predicted to interact with fluoxetine are to be administered soon after its discontinuation (see General: Implications of the Long Elimination Half-Life of Fluoxetine under Precautions; Drug-Drug Interactions: Tricyclic Antidepressants under Interactions).
Kinetic Data: After 30 days of dosing at 20 mg/day, mean plasma concentrations of fluoxetine 79.1 ± 33.4 ng/mL and of norfluoxetine 129 ± 42.0 ng/mL have been observed. Plasma concentrations of fluoxetine (elimination half-life of 1 to 3 days after acute administration and 4 to 6 days after chronic administration) were higher than those predicted by single-dose studies. Norfluoxetine appears to have linear pharmacokinetics. Its mean terminal half-lives after a single dose and multiple doses were 8.6 days and 9.3 days, respectively.
Steady state plasma levels are attained after 4 to 5 weeks of continuous drug administration. Patients receiving fluoxetine at doses of 40 to 80 mg/day over periods as long as 3 years exhibited, on average, plasma concentrations similar to those seen among patients treated for 4 to 5 weeks at the same dose.
Protein Binding: Approximately 94% of fluoxetine is protein bound. The interaction between fluoxetine and other highly protein bound drugs has not been fully evaluated, but may be important (see Drug-Drug Interactions: Drugs Tightly Bound to Plasma Protein under Interactions).
Special Populations and Conditions: Age: The effects of age upon the metabolism of fluoxetine have been investigated in a subset of 260 elderly, but otherwise healthy, depressed patients (mean age: 67.4 yr, range 60 to 85 yr) who received 20 mg fluoxetine for 6 weeks. Mean plasma concentrations were found to be 89.5 ± 53.6 ng/mL for fluoxetine and 119 ± 51.3 ng/mL for norfluoxetine. However, the effects of concomitant illness and/or concomitant drugs have not been evaluated.
Hepatic Insufficiency: In patients with cirrhosis, the elimination half-life of fluoxetine was prolonged, with a mean of 7.6 days compared to a range of 2 to 3 days seen in healthy subjects; norfluoxetine half-life was also prolonged, with a mean of 12 days compared to a range of 7 to 9 days in healthy subjects. Fluoxetine should therefore be used with caution in patients with liver disease (see Special Patient Populations: Debilitated Patients under Dosage & Administration; Hepatic/Biliary/Pancreatic: Hepatic Impairment under Precautions).
Renal Insufficiency: In single dose studies, the pharmacokinetics of fluoxetine and norfluoxetine were similar among subjects with all levels of impaired renal function including anephric patients on chronic hemodialysis. However, with chronic administration, additional accumulation of fluoxetine or its metabolites (possibly including some not yet identified) may occur in patients with severely impaired renal function, and the use of a lower or less frequent dose is advised (see Special Patient Populations: Debilitated Patients under Dosage & Administration; Renal: Severe Renal Impairment under Precautions).
Animal and In vitro: Fluoxetine was well absorbed orally and the oral bioavailability of fluoxetine in dogs was 72%. In dogs given oral doses of 1 to 10 mg/kg fluoxetine for one year, dose dependent increases in fluoxetine and norfluoxetine concentrations were observed in liver, adrenal, and lung.
Norfluoxetine concentrations exceeded fluoxetine concentrations in the tissues, and persisted for a longer period in plasma.
In rats, after a single i.p. dose of 10 mg/kg, the plasma half-life of fluoxetine was 26 hours and that of norfluoxetine, 40 hours. The plasma half-life in dogs dosed orally at 5 to 10 mg/kg for 15 days, was 1 day for fluoxetine and 2.1 to 5.4 days for norfluoxetine.
In vitro, fluoxetine was N-demethylated to norfluoxetine by rat, guinea pig, and rabbit liver microsomes. In vivo, fluoxetine was metabolized mainly by N-demethylation in mice, rats, guinea pigs, rabbits, and dogs. The other major metabolite was trifluoro-methylphenol, formed by O-dealkylation, which was excreted as a sulphate or glucuronide conjugate by rats, guinea pigs, and dogs.
Fluoxetine and norfluoxetine were also excreted in the urine unchanged in guinea pigs, rabbits, and dogs. In rats, fluoxetine and norfluoxetine were both further metabolized, so that neither fluoxetine nor its N-demethylated metabolite was found in the urine. Rats eliminated 16 to 42 percent of the dose in urine as p-trifluoromethylphenol and 8 percent of the dose as hippuric acid in 24 hours.
Safety Pharmacology: In vitro studies have demonstrated that fluoxetine and norfluoxetine inhibit hERG potassium channel and L-type calcium channel, as well as trafficking of the hERG channel to the plasma membrane.
Toxicology: Acute Toxicity Studies: See Table 3.
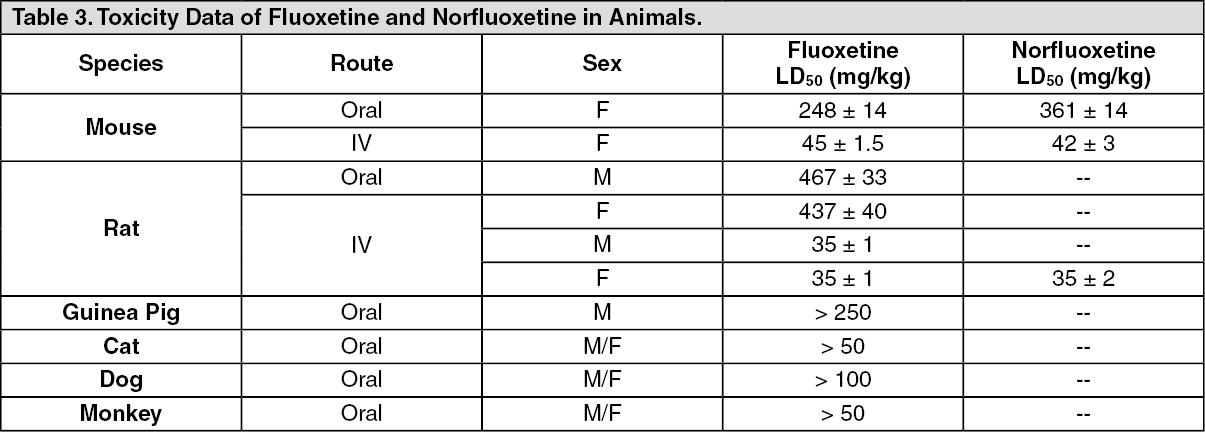 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageSigns of toxicity included vomiting, anorexia, mydriasis, salivation, tremors, clonic convulsions, hyperirritability and cachexia.
Subchronic/Chronic/Carcinogenicity and Related Toxicity Studies: Subchronic Toxicity Studies: Mice (5/sex/dose) were maintained on diets containing ca. 25, 59 and 204 mg/kg/day fluoxetine for 15 days. Thirty and 100% mortality were observed at the middle and high dose, respectively. Significant effects at the two highest doses included: hyperactivity and body weight loss, decrease in spleen weights and phospholipidosis.
Mice were maintained for three months on diets equivalent to ca. 2, 7 or 31 mg/kg/day. Significant effects were essentially limited to high dose mice and included 15% mortality; persistent hyperactivity and decreased body weight gain; slight and reversible increases in alkaline phosphatase and alanine transaminase; decreases in testes, heart, and spleen weights; hypospermatogenesis; reversible pulmonary phospholipidosis.
Pulmonary histiocytosis (phospholipidosis) was the major pathological finding in rats maintained on diets providing average doses of approximately 9, 25 or 74 mg/kg/day for three months. All animals at ca. 74 mg/kg/day died by week 8. Decreased food consumption, weight loss, and hyperirritability were observed at ca. 25 and 74 mg/kg/day.
Dogs given 5 to 50 mg/kg/day orally for two weeks experienced anorexia, mydriasis and vomiting. Dogs, receiving 50 mg/kg/day exhibited ataxia, tremors and a convulsion in one dog.
Dogs survived oral doses up to 20 mg/kg/day for three months with significant anorexia as the major treatment-related effect. Significant accumulation of both fluoxetine and norfluoxetine occurred in the plasma and tissues. Mydriasis and tremors were observed during the first month.
Monkeys given 10 or 25 mg/kg/day p.o. for two weeks exhibited anorexia and weight loss. One monkey at 25 mg/kg/day exhibited clonic convulsions after six doses. Accumulation of fluoxetine and norfluoxetine was observed after multiple dosing and decreased erythrocyte and white blood cell counts were observed.
Chronic Toxicity Studies: Fluoxetine was given daily to rats (25/sex/dose) for one year at dietary levels of ca. 0.5, 2.3 and 10.7 mg/kg/day. Physical signs of toxicity were limited to females at the high dose level and consisted of anorexia, chromodacryorrhea and an unusual behaviour first noted during the eighth month of treatment in which the animals walked on their toes with feet extended and backs arched after they had been handled.
Evidence of phospholipidosis was obtained in the lung, liver and adrenal cortex of 24/40 animals at the high dose level and in one rat at the mid-dose level. Phospholipidosis was reversible after two months' withdrawal from treatment. Minimal to slight fat deposition in the liver was prevalent at the mid and high dose levels. Reversible, minimal reticuloendothelial cell hyperplasia was present in the lymph nodes of the high dose level animals.
Dogs (5/sex/dose) received daily oral doses of 1, 4.5, or 20 mg/kg (decreased to 10 mg/kg after 6 months as three females died) of fluoxetine for one year. The toxic effects observed in this study were similar to those of the subchronic study except that phospholipidosis was seen after chronic administration in the lung, liver, adrenals, the inner plexiform layer of the retina, lymph nodes, spleen, and peripheral leukocytes in the animals receiving the high dose. They also showed moderate bradycardia and a moderate decrease in adrenal weight.
Phospholipidosis was only observed in the lung and leukocytes in a few of the dogs at the lowest dose level of 1.0 mg/kg/day. No cardiovascular effects were seen apart from a slight decrease in basal heart rate. All treatment-related effects were reversible during the recovery period in surviving animals.
Discussion on Phospholipidosis: Systemic phospholipidosis was associated with the subchronic and/or chronic administration of fluoxetine to mice, rats and dogs. This effect was associated with the accumulation of norfluoxetine, and to a lesser extent, fluoxetine, in affected tissues. Systemic phospholipidosis was not associated with any adverse effects and was shown to be reversible after the chronic administration of fluoxetine for one year in rats and dogs.
This effect has been demonstrated in animals with a number of other clinically useful cationic amphiphilic drugs including antidepressants - imipramine, clomipramine, iprindole and other drugs - chlorphentermine, fenfluramine, clozapine, chloroquine, mepacocine, chlorcyclizine, tamoxifen, 4,4'diethylaminoethoxyhexestrol, amiodarone and perhexiline. The significance of this finding for man is not fully understood. It is anticipated that in the clinical use of fluoxetine, the properties of the drug which are associated with phospholipidosis will not result in any untoward effect.
Juvenile Toxicology Study: In a juvenile toxicology study in CD rats, administration of 30 mg/kg of fluoxetine hydrochloride on postnatal days 21 through 90 resulted in increased serum activities of creatine kinase (CK) and aspartate aminotransferase (AST), which were accompanied microscopically by skeletal muscle degeneration, necrosis and regeneration. Femur lengths at 30 mg/kg increased to a lesser extent compared with control rats. The dose of 30 mg/kg was associated with severe toxicity in general and exceeded a maximum tolerated dose. A specific effect of fluoxetine on bone development has been reported in mice treated with fluoxetine during the juvenile period. When mice were treated with fluoxetine (5 or 20 mg/kg, ip) for 4 weeks starting at 4 weeks of age, bone formation was reduced resulting in decreased bone mineral content and density. These doses did not affect overall growth (body weight gain or femoral length). The doses administered to juvenile mice in this study are approximately 0.5 and 2 times the MRD for pediatric patients on a body surface basis. Other findings are discussed as follows in the Reproductive and Impairment of Fertility Studies.
Carcinogenicity Studies: Rats were maintained for two years at dietary levels equivalent to a time-weighted average dose of ca. 0.45, 2 and 9 mg/kg/day. Age-related observations such as chromodacryorrhea, alopecia, and poor grooming increased at the high dose, especially in females. Weight gain and food consumption were depressed at the high dose and a handling-induced behaviour involving arching of the back and walking on toes was observed primarily in females in this group.
Increased tissue levels of fluoxetine and norfluoxetine were observed at all doses, and phospholipidosis was observed primarily at the high dose. There were no significant increases in tumor incidence or animal mortality.
Mice were fed dietary levels of fluoxetine equivalent to ca. 1.2, 4.8 and 12.1 mg/kg/day. The dietary levels were based on the results of the three-month subchronic study. Unexpectedly, high mortality occurred in females receiving the high dose early in the two-year study, necessitating lowering the dose after 30 days. The survival rate of females receiving the high dose was reduced at two years. No major toxicological effects were seen in mice other than a moderate increase in alanine transaminase in males receiving the high dose and slight changes in organ weights. Hepatocellular degeneration, fat deposition in liver, and centrilobular hepatocellular degeneration were observed microscopically at the median and high dose. There was no evidence of phospholipid accumulation in the lung, and no oncogenic response was observed.
A second two-year mouse study using similar doses gave similar results. Survival at two years was reduced in females receiving the high dose. Handling-induced clonic convulsions occurred at all levels in males, and in females, at the high-dose level it was accompanied by a slight increase in liver weight. Minimal-to-moderate fatty change in the liver and hepatocellular cytomegaly were seen in mice from the median- and high-dose levels. There was a dose-dependent increase in concentrations of fluoxetine and norfluoxetine in lung tissue. There was no evidence of phospholipid accumulation in the lung, and no oncogenic response was observed.
Reproductive and Impairment of Fertility Studies: Female Wistar rats (30/dose) were given daily oral doses of 2, 5, or 12.5 mg/kg from two weeks prior to mating through gestation or lactation. In a second study, male Wistar rats (40/dose) were maintained on diets approximately equivalent to 1.5, 3.9, or 9.7 mg/kg for 10 weeks prior to mating and through the breeding trial. These treated males were mated with female Wistar rats (40/dose) maintained at the same dietary levels for three weeks prior to mating and throughout lactation. In both studies, a depression in neonatal survival was obtained at the high dose level.
No teratogenic effects or adverse effects on fertility or post-natal development were associated with fluoxetine administration.
Impairment of fertility in adult animals at doses up to 12.5 mg/kg/day (approximately 1.5 times the MRHD on a mg/m2 basis) was not observed.
In a juvenile toxicology study, fluoxetine hydrochloride was administered orally to CD rats (30/sex/group) at doses of 0, 3, 10, and 30 mg/kg/day from postnatal days 21 through 91 and evaluated for general clinical observations. Ten rats/sex/group were necropsied on postnatal day 91 and evaluated for changes in clinical chemistry, hematology, femur length, organ weights, and histopathology. Following an approximately 11-week recovery period, sperm assessments were performed in all groups, and microscopic examination of testis and epididymides occurred in the 30 mg/kg/day males only.
Plasma levels achieved at 30 mg/kg/day were approximately 5 to 8 fold (fluoxetine) and 18 to 20 fold (norfluoxetine), and at 10 mg/kg approximately 2 fold (fluoxetine) and 8 fold (norfluoxetine) higher compared to plasma concentrations usually achieved in pediatric patients.
Administration of 30 mg/kg/day of fluoxetine hydrochloride on postnatal days 21 through 90 resulted in a substantial decrease in body weight gain with concomitant degeneration and necrosis of seminiferous tubules of the testis, epididymal epithelial vacuolation, epididymal sperm granuloma, and immaturity and inactivity of the female reproductive tract.
Findings following an approximately 11-week recovery period in male rats administered 30 mg/kg/day, consisted of testicular degeneration, seminiferous tubular sperm microgranulomas, epididymal epithelial cribriform change, epididymal epithelial vacuolation and epididymal sperm granulomas. All of the rats with cribriform change had testicular degeneration, and comparison to the treatment-phase rats indicted that the testicular degeneration was irreversible. In contrast, the reduction in degree and extent of epididymal vacuolation compared to the treatment-phase rats indicates that the vacuolation was reversible.
Sperm assessments in the 30-mg/kg males only indicated an approximately 30% decrease in sperm concentrations without affecting sperm morphology or motility. Decreased fertility was observed in this dose-group. Delays in sexual maturation occurred in the 10-mg/kg/day males and in the 30 mg/kg/day males and females. The significance of these findings in humans is unknown.
Mutagenicity Studies: The mutagenicity of fluoxetine and its metabolite norfluoxetine was evaluated in a battery of in vitro and in vivo tests including Ames test, modified Ames test, DNA repair in rat hepatocytes, sister chromatid exchange in Chinese hamster bone marrow assays, and mouse lymphoma assay. Fluoxetine and norfluoxetine were negative in all 5 systems.
Teratology Studies: Virgin female Fischer 344 rats (25/dose) were bred with untreated control males and were given daily oral (gavage) doses of 2, 5, or 12.5 mg/kg/day fluoxetine on gestation days 6-15; animals were evaluated on gestation day 20. Body weight gains and food consumption were depressed at 12.5 mg/kg/day. Fluoxetine produced no teratogenic effects and no changes in reproductive parameters.
Virgin female Dutch Belted rabbits (15/dose) were artificially inseminated with semen from untreated control males and were given daily oral (gavage) doses of 2.5, 7.5, or 15 mg/kg/day fluoxetine on gestation days 6-18; animals were evaluated on gestation day 28. Maternal toxicity was demonstrated by depressed body weight gains and food consumption at all dose levels in a dose-dependent manner. At the 15 mg/kg/day dose, two rabbits died and three aborted. Resorptions were also increased in this group. There was no evidence of a teratogenic effect.