One ml contains: Human normal immunoglobulin 100 mg (purity of at least 98% IgG).
Each vial of 50 ml solution contains: 5 g human normal immunoglobulin.
Distribution of the IgG subclasses (approx. values): IgG1 69%; IgG2 26%; IgG3 3%; IgG4 2%.
The maximum IgA content is 25 micrograms/ml.
Produced from the plasma of human donors.
Privigen is isotonic, with an approximate osmolality of 320 mOsmol/kg.
Excipients with known effects: Privigen contains approximately 250 mmol/L (range: 210 to 290) of L-proline.
Excipients/Inactive Ingredients: L-proline, Water for injections, Hydrochloric acid (for pH adjustment), Sodium hydroxide (for pH adjustment).
Pharmacotherapeutic group: immune sera and immunoglobulins: immunoglobulins, normal human, for intravascular administration.
ATC code: J06BA02.
Pharmacology: Pharmacodynamics: Human normal immunoglobulin contains mainly immunoglobulin G (IgG) with a broad spectrum of antibodies against infectious agents.
Human normal immunoglobulin contains the IgG antibodies present in the normal population. It is usually prepared from pooled plasma from not fewer than 1,000 donors. It has a distribution of immunoglobulin G subclasses closely proportional to that in native human plasma. Adequate doses of this medicinal product may restore abnormally low immunoglobulin G levels to the normal range
and thus help against infections.
The mechanism of action in indications other than replacement therapy is not fully elucidated, but includes immunomodulatory effects.
The safety and efficacy of Privigen was
evaluated in 7 prospective, open-label, single-arm, multicenter studies performed in Europe (ITP, PID and CIDP studies), Japan (PID and CIDP studies) and the US (PID and CIDP studies).
Additional safety data were collected in a Post-Authorization Safety Study (PASS), an observational multicentre trial in patients with various immunological conditions performed in the US.
PID: The PID pivotal study included a total of 80 patients aged between 3 and 69 years old.
19 children (3 to 11 years), 12 adolescents (12 to 16 years) and 49 adults were treated with Privigen over 12 months. 1038 infusions were administered, 272 (in 16 patients) in the 3-week schedule and 766 (in 64 patients) in the 4-week schedule. The median doses administered for the 3-week and 4-week treatment schedules were almost identical to each other (428.3 vs. 440.6 mg IgG/kg bw). The PID extension study included a total of 55 patients
aged between 4 and 81 years old. 13 children (3 to 11 years), 8 adolescents (12 to 15 years) and 34 adults were treated with Privigen over 29 months. 771 infusions were administered and the median dose administered was 492.3 mg IgG/kg bw.
ITP: In the ITP pivotal study, in total 57 patients aged between 15 and 69 years old
were treated with 2 infusions of Privigen for a total of 114 infusions. The scheduled dose of 1 g/kg bw per infusion was closely adhered to in all patients (median 2 g IgG/kg bw).
In the second ITP study, 57 patients with ITP (baseline platelet counts ≤30×109/l) aged between 18 and 65 years were treated with Privigen at 1 g/kg bw. On day 3 patients could receive a second dose of 1 g/kg bw, for patients with a platelet count of <50×109/l on day 3 this second dose was mandatory. Overall, in 42 subjects (74%) the platelet count increased at least once to ≥50×109/l within 6 days after the first infusion, which was well within the expected range. A second dose in subjects with platelet counts ≥50×109/l after the first dose provided a relevant additional benefit in terms of higher and longer-lasting increases in platelet counts compared to a single dose. In subjects with platelet counts <50×109/l after the first dose, 30% showed a platelet response of ≥50×109/l after the mandatory second dose.
CIDP: In the first CIDP study, a prospective multicenter open label trial (Privigen impact on mobility and autonomy PRIMA study), 28 patients (13 subjects who have previously received IVIG and 15 subjects not) were treated with a Privigen loading dose of 2 g/kg bw given over 2-5 days followed by 6 maintenance doses of 1 g/kg bw over 1-2 days every three weeks. Previously treated patients were withdrawn from IVIG until confirmed deterioration before start of Privigen. On the adjusted 10-point INCAT (Inflammatory Neuropathy Cause and Treatment) scale a clinically meaningful improvement of at least 1-point from baseline to treatment week 25 was observed in 17 out of 28 patients. The INCAT responder rate was 60.7% (95% confidence interval [42.41, 76.4]). 9 patients responded after receiving the initial induction dose by week 4, 16 patients responded by week 10.
Muscle strength as measured by the MRC (Medical Research Council) Score improved in all patients by 6.9 points (95% confidence interval [4.11, 9.75], in previously treated patients by 6.1 points (95% confidence interval [2.72, 9.44]) and in untreated patients by 7.7 points (95% confidence interval [2.89, 12.44]). The MRC responder rate, an increase of at least 3 points, was 84.8% which was similar in previously treated (81.5% [58.95, 100.00]) and untreated (86.7% [69.46, 100.00]) patients. In patients defined as INCAT non-responders, muscle strength improved by 5.5 points (95% confidence interval [0.6, 10.2]) as compared to INCAT responders (7.4 points (95% confidence interval [4.0, 11.7]).
In a second prospective, multicenter randomized, placebo-controlled clinical study (Polyneuropathy and Treatment with Hizentra, PATH trial), 207 subjects with CIDP were treated with Privigen in the prerandomization phase of the study. Subjects all with IVIg pretreatment of at least 8 weeks and with an IVIg-dependence confirmed by clinically evident deterioration during an IVIg withdrawal phase of up to 12 weeks, received a Privigen loading dose of 2 g/kg bw followed by up to 4 Privigen maintenance doses of 1 g/kg bw every 3 weeks for up to 13 weeks.
Following clinical deterioration during IVIg withdrawal, clinical improvement of CIDP was primarily defined by a decrease of ≥1 point at the adjusted INCAT score. Additional measures of CIDP improvement were an increase in R-ODS (Rasch-built Overall Disability Scale) score of ≥4 points, a mean grip strength increase of ≥8 kPa, or an MRC sum score increase of ≥3 points. Overall, 91% of subjects (188 patients) showed improvement in at least one of the previous criteria by week 13. By adjusted INCAT score, the responder rate by week 13 was 72.9% (151/207 patients), with 149 patients responding already by week 10. A total of 43 of the 207 patients achieved a better CIDP status as assessed by the adjusted INCAT score compared to their CIDP status at study entry.
The mean improvement at the end of the treatment period compared to reference visit was 1.4 points in the PRIMA (1.8 points in IVIg-pretreated subjects) and 1.2 points in PATH study.
In PRIMA, the percentage of responders in the overall Medical Research Council (MRC) score (defined as an increase by ≥3 points) was 85% (87% in the IVIg-untreated and 82% in IVIg-pretreated) and 57% in PATH. The overall median time to first MRC sum score response in PRIMA was 6 weeks (6 weeks in the IVIg-untreated and 3 weeks in the IVIg-pretreated) and 9.3 weeks in PATH. MRC sum score in PRIMA improved by 6.9 points (7.7 points for IVIg-untreated and 6.1 points for IVIg-pretreated) and by 3.6 points in PATH.
The grip strength of the dominant hand improved by 14.1 kPa (17.0 kPa in IVIg-untreated and 10.8 kPa in IVIg-pretreated subjects) in the PRIMA study, while in PATH the grip strength of the dominant hand improved by 12.2 kPa. For the non dominant hand similar results were observed in both PRIMA and PATH trials.
The efficacy and safety profile in the PRIMA and the PATH study in CIDP patients were overall comparable.
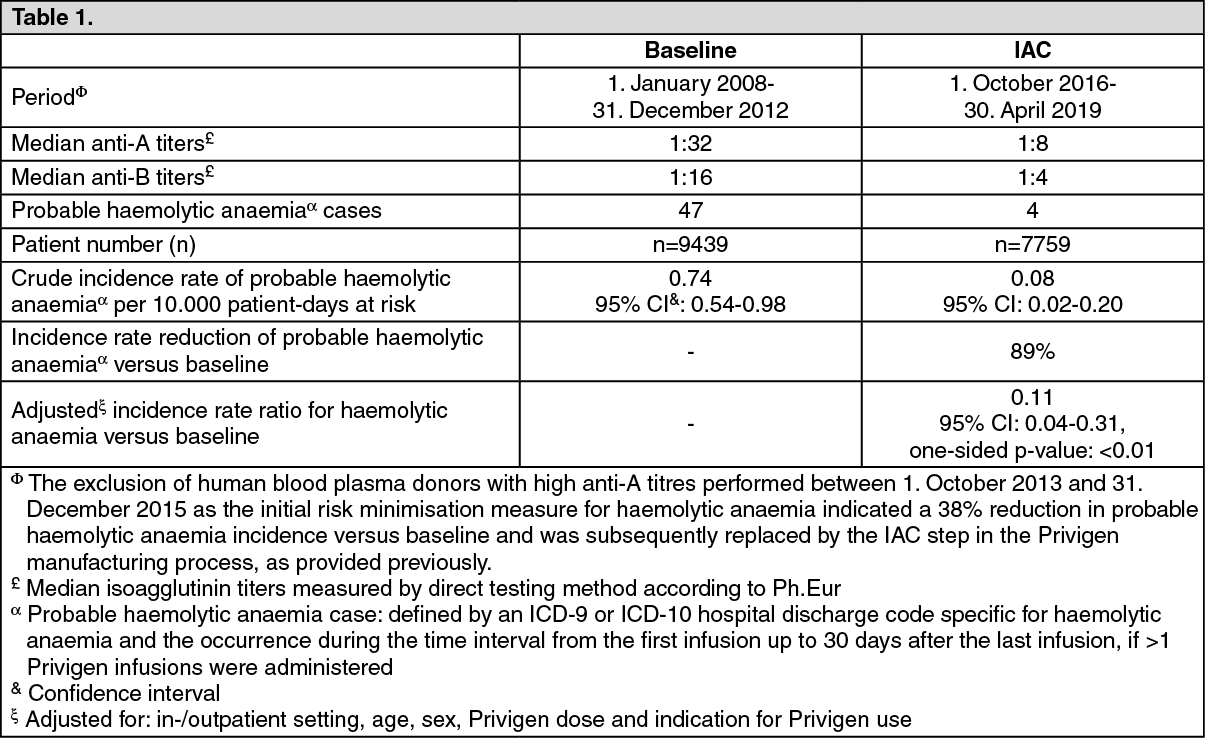
Post-Authorisation Safety Study (PASS): In an observational hospital-based cohort Post-Authorisation Safety Study (PASS), the risk of haemolytic anaemia following Privigen therapy was evaluated in patients with various immunological conditions from 1 January 2008 to 30 April 2019. The risk of haemolytic anaemia was assessed prior (baseline) and after the implementation of a risk minimisation measure, the introduction of the Immunoaffinity Chromatography (IAC) step in the Privigen manufacturing process. Probable cases of haemolytic anaemia were defined by an ICD-9 or ICD-10 hospital discharge code specific for haemolytic anaemia. (Possible cases of haemolytic anaemia consisted of an unspecified transfusion reaction identified via ICD-9 or ICD-10 discharge codes or via review of hospital charge descriptions in temporal association with a haptoglobin, a direct antiglobulin test or indirect antiglobulin performed in the workup of haemolytic anaemia).
A statistically significant rate reduction of 89% of haemolytic anaemia (based on an incidence rate ratio of 0.11; adjusted for in/outpatient setting, age, sex, Privigen dose and indication for Privigen use; one-sided p-value <0.01) was observed after implementation of the IAC step compared to baseline: See Table 1.
 Click on icon to see table/diagram/image
The reduction in probable haemolytic anaemia incidence rate after IAC implementation versus baseline was especially pronounced in patients treated with Privigen doses ≥0.75 g/kg bw.
Click on icon to see table/diagram/image
The reduction in probable haemolytic anaemia incidence rate after IAC implementation versus baseline was especially pronounced in patients treated with Privigen doses ≥0.75 g/kg bw.
Additionally, 28 paediatric patients with CIDP <18 years of age were identified throughout the entire study period from 1 January 2008 to 30 April 2019. No paediatric patients with CIDP given a total of 486 Privigen administrations experienced haemolytic anaemia, AMS, acute renal failure, severe anaphylactic reaction or a thromboembolic event. Two patients experienced a moderate anaphylactic reaction, equating to 0.4% of all Privigen administrations.
Paediatric population: No differences were observed in the pharmacodynamic properties and safety profile between adult and paediatric study patients.
Pharmacokinetics: Absorption: Human normal immunoglobulin is immediately and completely bioavailable in the recipient's circulation after intravenous administration.
Distribution: It is distributed relatively rapidly between plasma and extravascular fluid, after approximately 3-5 days equilibrium is reached between the intra- and extravascular compartments.
Elimination: IgG and IgG complexes are broken down in the cells of the reticuloendothelial system. The half-life may vary from patient to patient. The pharmacokinetic parameters for Privigen were determined in a clinical study in PID patients (see Pharmacodynamics as previously mentioned). 25 patients (aged 13-69 years) participated in the pharmacokinetic (PK) assessment. In this study, the median half-life of Privigen in PID patients was 36.6 days. In an extension of this study, 13 PID patients
(aged 3-65 years) participated in a PK sub-study. The results of this study show the median half-life of Privigen to be 31.1 days
(see Table 2).
Click on icon to see table/diagram/image
Paediatric population: No differences were seen in the pharmacokinetic parameters between adult and paediatric study patients with PID. There are no data on pharmacokinetic properties in paediatric patients with CIDP.
Toxicology: Preclinical safety data: Immunoglobulins are a normal constituent of the human body. L-proline is a physiological, non-essential amino acid.
The safety of Privigen has been assessed in several preclinical studies, with particular reference to the excipient L-proline. Some published studies pertaining to hyperprolinaemia have shown that long-term, high doses of L-proline have effects on brain development in very young rats. However, in studies where the dosing was designed to reflect the clinical indications for Privigen, no effects on brain development were observed.
Non-clinical data reveal no special risk for humans based on safety pharmacology and toxicity studies.
Replacement therapy in adults, and children and adolescents (0-18 years) in: Primary immunodeficiency syndromes (PID) with impaired antibody production (see Precautions); Secondary immunodeficiencies (SID) in patients who suffer from severe or recurrent infections, ineffective antimicrobial treatment and either proven specific antibody failure (PSAF)* or serum IgG level of <4 g/l.
* PSAF = failure to mount at least a 2-fold rise in IgG antibody titre to pneumococcal polysaccharide and polypeptide antigen vaccines.
Immunomodulation in adults, and children and adolescents (0-18 years) in: Primary immune thrombocytopenia (ITP), in patients at high risk of bleeding or prior to surgery to correct the platelet count; Guillain-Barré syndrome; Kawasaki disease (in conjunction with acetylsalicylic acid; see Dosage & Administration); Chronic inflammatory demyelinating polyneuropathy (CIDP) - only limited experience is available of use of intravenous immunoglobulins in children with CIDP; Multifocal motor neuropathy (MMN).
Replacement therapy should be commenced and monitored under the supervision of a physician experienced in the treatment of immunodeficiency.
Posology: The dose and dose regimen is dependent on the indication.
In replacement therapy, the dose may need to be individualised for each patient depending on the clinical response.
Dose based on body weight may require adjustment in underweight or overweight patients. The following dose regimens are given as a guideline.
Replacement therapy in primary immunodeficiency (PID) syndromes: The dose regimen should achieve a trough IgG level (measured before the next infusion) of at least
6 g/l or within the normal reference range for the population age. Three to six months are required after the initiation of therapy for equilibration to occur. The recommended starting dose is 0.4 to 0.8 g/kg body weight (bw)
given once, followed by at least 0.2 g/kg bw every
3 to 4 weeks.
The dose required to achieve a trough level of IgG of 6 g/l is of the order of 0.2 to 0.8 g/kg bw/month. The dosage interval when steady state has been reached varies from
3 to 4 weeks.
IgG trough levels should be measured
and assessed in conjunction with the incidence of infection. To reduce the rate of bacterial infections, it may be necessary to increase the dosage and aim for higher trough levels.
Secondary immunodeficiencies (as defined in Indications/Uses): The dose regimen should achieve a trough IgG level (measured before the next infusion) of at least 6 g/l or within the normal reference range for the population age. The recommended dose is 0.2-0.4 g/kg bw every three to four weeks.
IgG trough levels should be measured and assessed in conjunction with the incidence of infection. Dose should be adjusted as necessary to achieve optimal protection against infections, an increase may be necessary in patients with persisting infection; a dose decrease can be considered when the patient remains infection free.
Primary immune thrombocytopenia (ITP): There are two alternative treatment schedules: 0.8 to 1 g/kg bw given on day 1 - this dose may be repeated once within 3 days; 0.4 g/kg bw given daily for 2 to 5 days.
The treatment can be repeated if relapse occurs.
Guillain-Barré syndrome: 0.4 g/kg bw/day over 5 days
(possible repeat of dosing in case of relapse).
Kawasaki disease: 2.0 g/kg bw should be administered as a single dose.
Patients should receive concomitant treatment with acetylsalicylic acid.
Chronic inflammatory demyelinating polyneuropathy (CIDP)*: The recommended starting dose is 2 g/kg bw divided over 2 to 5 consecutive days followed by maintenance doses of 1 g/kg bw over 1 to 2 consecutive days every 3 weeks.
The treatment effect should be evaluated after each cycle; if no treatment effect is seen after 6 months, the treatment should be discontinued.
If the treatment is effective, long-term treatment should be subject to the physician's discretion based upon the patient response and maintenance response. The dosing and intervals may have to be adapted according to the individual course of the disease.
Multifocal Motor Neuropathy (MMN): Starting dose: 2 g/kg given over 2-5 consecutive days.
Maintenance dose: 1 g/kg every 2 to 4 weeks or 2 g/kg every 4 to 8 weeks.
The treatment effect should be evaluated after each cycle. If insufficient treatment effect is seen after 6 months, the treatment should be discontinued.
If the treatment is effective, long-term treatment should be subject to the physician's discretion based upon the patient response. The dosing and intervals may have to be adapted according to the individual course of the disease.
The dosage recommendations are summarised in the following table:
See Table 3.
Click on icon to see table/diagram/image
Paediatric population: The posology in children and adolescents (0-18 years) is not different from that of adults as the posology for each indication is given by body weight and adjusted to the clinical outcome of the previously mentioned conditions.
Hepatic impairment: No evidence is available to require a dose adjustment.
Renal impairment: No dose adjustment unless clinically warranted, see Precautions.
Elderly: No dose adjustment unless clinically warranted, see Precautions.
Method of administration: For intravenous use.
Privigen should be infused intravenously at an initial infusion rate of 0.3 ml/kg bw/hr for approximately 30 min. If well tolerated (see Precautions), the rate of administration may gradually be increased to 4.8 ml/kg bw/hr.
In PID patients who have tolerated the infusion rate of 4.8 ml/kg bw/hr well, the rate may be further gradually increased to a maximum of 7.2 ml/kg bw/hr.
If dilution prior to infusion is desired, Privigen may be diluted with 5% glucose solution to a final concentration of 50 mg/ml (5%). For instruction, see Special precautions for disposal and other handling under Cautions for Usage.
Overdose may lead to fluid overload and hyperviscosity, particularly in patients at risk, including elderly patients or patients with cardiac or renal impairment.
Hypersensitivity to the active substance (human immunoglobulins) or to any of the excipients listed in Description (see also Precautions).
Patients with selective IgA deficiency who developed antibodies to IgA, as administering an IgA-containing product can result in anaphylaxis.
Patients with hyperprolinaemia type I or II.
Traceability: In order to improve the traceability of biological medicinal products, the name and the batch number of the administered product should be clearly recorded.
Certain severe adverse reactions may be related to the rate of infusion. The recommended infusion rate given under Dosage & Administration must be closely followed. Patients must be closely monitored and carefully observed for any symptoms throughout the infusion period.
Certain adverse reactions may occur more frequently: in case of high rate of infusion; in patients with hypogammaglobulinaemia or agammaglobulinaemia, with or without IgA deficiency; in patients who receive human normal immunoglobulin for the first time or, in rare cases, when the human normal immunoglobulin product is switched or when there has been a long interval since the previous infusion.
Potential complications can often be avoided by ensuring that patients: are not sensitive to human normal immunoglobulin by initially infusing the product slowly (0.3 ml/kg bw/hr); are carefully monitored for any symptoms throughout the infusion period. In particular, patients naïve to human normal immunoglobulin, patients switched from an alternative IVIg product or when there has been a long interval since the previous infusion should be monitored during the first infusion and for the first hour after the first infusion, in order to detect potential adverse signs. All other patients should be observed for at least 20 minutes after administration.
In case of adverse reaction, either the rate of administration must be reduced or the infusion stopped. The treatment required depends on the nature and severity of the adverse reaction.
In all patients, IVIg administration requires: adequate hydration prior to the initiation of the infusion of IVIg; monitoring of urine output; monitoring of serum creatinine levels; avoidance of concomitant use of loop diuretics (see Interactions).
For patients suffering from diabetes mellitus and requiring dilution of Privigen to lower concentrations, the presence of glucose in the recommended diluent should be taken into account.
Hypersensitivity: True hypersensitivity reactions are rare. They can occur in patients with anti-IgA antibodies.
IVIg is not indicated in patients with selective IgA deficiency where the IgA deficiency is the only abnormality of concern.
Rarely, human normal immunoglobulin can induce a fall in blood pressure with anaphylactoid reaction, even in patients who had tolerated previous treatment with human normal immunoglobulin. In case of shock, standard medical treatment for shock should be implemented.
Haemolysis: IVIg products can contain blood group antibodies which may act as haemolysins and induce in vivo coating of red blood cells (RBC) with immunoglobulin, causing a positive direct antiglobulin reaction (Coombs' test) and, rarely, haemolysis. Haemolytic anaemia can develop subsequent to IVIg therapy due to enhanced RBC sequestration. The Privigen manufacturing process includes an immunoaffinity chromatography (IAC) step that specifically reduces blood group A and B antibodies (isoagglutinins A and B). Clinical data with Privigen manufactured with the IAC step show statistically significant reductions of haemolytic anaemia (see Adverse Reactions and Actions).
Heightened awareness of the potential for haemolysis is recommended in individuals receiving normal immunoglobulin products, particularly those who are determined to be at increased risk.
Patients at increased risk for haemolysis following treatment with normal immunoglobulin include those with non-O blood group types, those who have underlying associated inflammatory conditions, and those receiving high cumulative doses of normal immunoglobulin over the course of several days.
The following risk factors are associated with the development of haemolysis: high doses, whether given as a single administration or divided over several days; non-O blood group; and underlying inflammatory state. As this event was commonly reported in non-O blood group patients receiving high doses for non-PID indications, increased vigilance is recommended. Haemolysis has rarely been reported in patients given replacement therapy for PID.
Patients receiving normal immunoglobulin products should be monitored for haemolysis, particularly those at increased risk.
Clinical symptoms and signs of haemolysis include fever, chills and dark urine. If these occur, appropriate laboratory testing should be obtained.
IVIg recipients should be monitored for clinical signs and symptoms of haemolysis. If signs and/or symptoms of haemolysis develop during or after an IVIg infusion, discontinuation of the IVIg treatment should be considered by the treating physician (see also Adverse Reactions).
Isolated cases of haemolysis-renal dysfunction/renal failure or disseminated intravascular coagulation and death have occurred.
Aseptic meningitis syndrome (AMS): Aseptic meningitis syndrome has been reported to occur in association with IVIg treatment. The syndrome usually begins within several hours to 2 days following IVIg treatment. Cerebrospinal fluid studies are frequently positive with pleocytosis up to several thousand cells per mm3, predominantly from the granulocytic series, and elevated protein levels up to several hundred mg/dl. AMS may occur more frequently in association with high-dose (2 g/kg) IVIg treatment. Patients exhibiting such signs and symptoms should receive a thorough neurological examination, including CSF studies, to rule out other causes of meningitis.
Discontinuation of IVIg treatment has resulted in remission of AMS within several days without sequelae.
Thrombosis: Care should be used when normal immunoglobulin products are given to individuals determined to be at increased risk of thrombosis.
Patients at increased risk of thrombosis include those with acquired or hereditary hypercoagulable states, prolonged immobilization, in-dwelling vascular catheters, advanced age, estrogen use, a history of venous or arterial thrombosis, cardiovascular risk factor (including history of atherosclerosis and/or impaired cardiac output), and hyperviscosity (including cryoglobulins, fasting chylomicronemia and/or high triglyceride levels, and monoclonal gammopathies).
Caution should be exercised in prescribing and infusing IVIg in obese patients.
Patients at risk for thrombosis should receive normal immunoglobulin products at the slowest infusion rate practicable, and these individuals should be monitored for thrombotic complications.
Consideration should also be given to measurement of baseline blood viscosity in individuals at risk of hyperviscosity.
There is clinical evidence of an association between IVIg administration and thromboembolic events such as myocardial infarction, cerebral vascular accident (including stroke), pulmonary embolism and deep vein thromboses which is assumed to be related to a relative increase in blood viscosity through the high influx of immunoglobulin in at-risk patients.
Caution should be exercised in prescribing and infusing IVIg in obese patients and in patients with pre-existing risk factors for thrombotic events (such as advanced age, hypertension, diabetes mellitus and a history of vascular disease or thrombotic episodes, patients with acquired or inherited thrombophilic disorders, patients with prolonged periods of immobilisation, severely hypovolaemic patients, patients with diseases which increase blood viscosity).
In patients at risk for thromboembolic adverse reactions, IVIg products should be administered at the minimum rate of infusion and dose practicable.
Acute renal failure: Cases of acute renal failure have been reported in patients receiving IVIg therapy. In most cases risk factors have been identified, such as pre-existing renal insufficiency, diabetes mellitus, hypovolaemia, overweight, concomitant nephrotoxic medicinal products or age over 65.
Renal parameters should be assessed prior to infusion of IVIg, particularly in patients judged to have a potential increased risk for developing acute renal failure, and again at appropriate intervals.
In case of renal impairment, IVIg discontinuation should be considered. While these reports of renal dysfunction and acute renal failure have been associated with the use of many of the licensed IVIg products containing various excipients such as sucrose, glucose and maltose, those containing sucrose as a stabiliser accounted for a disproportionate share of the total number. In patients at risk, the use of IVIg products that do not contain sucrose should therefore be considered. Privigen does not contain sucrose, maltose or glucose.
In patients at risk of acute renal failure, IVIg products should be administered at the minimum rate of infusion and dose practicable based on clinical judgement.
Transfusion-related acute lung injury (TRALI): In patients receiving IVIg, there have been some reports of acute non-cardiogenic pulmonary oedema [Transfusion Related Acute Lung Injury (TRALI)]. TRALI is characterised by severe hypoxia, dyspnoea, tachypnoea, cyanosis, fever and hypotension. Symptoms of TRALI typically develop during or within 6 hours of a transfusion, often within 1-2 hours. Therefore, IVIg recipients must be monitored for and IVIg infusion must be immediately stopped in case of pulmonary adverse reactions. TRALI is a potentially life-threatening condition requiring immediate intensive-care-unit management.
Interference with serological testing: After injection of immunoglobulin, the transitory rise of the various passively transferred antibodies in the patient's blood may result in misleading positive results in serological testing.
Passive transmission of antibodies to erythrocyte antigens, e.g. A, B, D, may interfere with some serological tests for red cell antibodies, for example the direct antiglobulin test (DAT, direct Coombs' test).
Transmissible agents: Privigen is made from human plasma. Standard measures to prevent infections resulting from the use of medicinal products prepared from human blood or plasma include selection of donors, screening of individual donations and plasma pools for specific markers of infection and the inclusion of effective manufacturing steps for the inactivation/removal of viruses. Despite this, when medicinal products prepared from human blood or plasma are administered, the possibility of transmitting infective agents cannot be totally excluded. This also applies to unknown or emerging viruses and other pathogens.
The measures taken are considered effective for enveloped viruses such as human immunodeficiency virus (HIV), hepatitis B virus (HBV), and hepatitis C virus (HCV) and for the non-enveloped viruses such as hepatitis A virus (HAV) and parvovirus B19.
There is reassuring clinical experience regarding the lack of hepatitis A or parvovirus B19 transmission with immunoglobulins and it is also assumed that the antibody content makes an important contribution to the viral safety.
Sodium content: This medicinal product contains less than 2.3 mg sodium per 100 ml, equivalent to 0.12% of the WHO recommended maximum daily intake of 2 g sodium for an adult.
Effects on ability to drive and use machines: Privigen has minor influence on the ability to drive and use machines, e.g. dizziness (see Adverse Reactions). Patients who experience adverse reactions during treatment should wait for these to resolve before driving or operating machines.
Use in Children: Although limited data is available, it is expected that the same warnings, precautions and risk factors apply to the paediatric population. In post marketing reports it is observed that IVIG high-dose indications in children, particularly Kawasaki disease, are associated with an increased reporting rate of haemolytic reactions compared to other IVIG indications in children.
Pregnancy: The safety of this medicinal product for use in human pregnancy has not been established in controlled clinical trials and therefore should only be given with caution to pregnant women and breast-feeding mothers. IVIg products have been shown to cross the placenta, increasingly during the third trimester. Clinical experience with immunoglobulins suggests that no harmful effects on the course of pregnancy, or on the foetus and the neonate are to be expected.
Experimental studies of the excipient L-proline carried out in animals found no direct or indirect toxicity affecting pregnancy, embryonal or foetal development.
Breast-feeding: Immunoglobulins are excreted into the milk and may contribute to protecting the neonate from pathogens which have a mucosal portal of entry.
Fertility: Clinical experience with immunoglobulins suggests that no harmful effects on fertility are to be expected.
Summary of the safety profile: Adverse reactions such as chills, headache,
dizziness, fever, vomiting, allergic reactions, nausea, arthralgia, low blood pressure and
moderate low back pain may occur occasionally in connection with intravenous administration of human immunoglobulin.
Rarely human normal immunoglobulins may cause a sudden fall in blood pressure and, in isolated cases, anaphylactic shock, even when the patient has shown no hypersensitivity to previous administration.
Cases of reversible aseptic meningitis and rare cases of transient cutaneous reactions
(including cutaneous lupus erythematosus - frequency unknown) have been observed with human normal immunoglobulin.
Reversible haemolytic reactions have been observed in patients, especially those with blood groups A, B, and AB in immunomodulatory treatment. Rarely, haemolytic anaemia requiring transfusion may develop after high dose IVIg treatment (see Precautions).
Increase in serum creatinine level and/or acute renal failure have been observed.
Very rarely:
Transfusion related acute lung injury (TRALI) and thromboembolic reactions such as myocardial infarction, stroke, pulmonary embolism and deep vein thromboses.
Tabulated list of adverse reactions: Seven clinical studies were performed with Privigen, which included patients with PID, ITP and CIDP. In the pivotal PID study, 80 patients were enrolled and treated with Privigen. Of these, 72 completed the 12 months of treatment. In the PID extension study, 55 patients were enrolled and treated with Privigen.
Another clinical study included 11 PID patients in Japan. The two ITP studies were performed with 57 patients each. Two CIDP studies were performed with 28 and 207 patients, respectively.
Most adverse drug reactions (ADRs) observed in the
seven clinical studies were mild to moderate in nature.
The following table shows an overview of the ADRs observed in the
seven clinical studies categorized according to the MedDRA System Organ Class (SOC),
Preferred Term Level (PT) and frequency. Frequencies were evaluated according to the following conventions: Very common (≥1/10), Common (≥1/100 to <1/10), Uncommon (≥1/1,000 to <1/100),
Rare (≥1/10,000 to <1/1,000), Very rare (<1/10,000).
For spontaneous post-marketing ADRs, the reporting frequency is categorized as unknown.
Within each frequency grouping, undesirable effects are presented in order of decreasing frequency.
(See Table 4.)
Click on icon to see table/diagram/image
For safety with respect to transmissible agents
and additional details on risk factors, see Precautions.
Paediatric population: In Privigen clinical studies with paediatric patients, the frequency, nature and severity of adverse reactions did not differ between children and adults. In post marketing reports it is observed that the proportion of haemolysis cases to all case reports occurring in children is slightly higher than in adults. Please refer to Precautions for details on risk factors and monitoring recommendations.
Reporting of suspected adverse reactions: Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions.
Live attenuated virus vaccines: Immunoglobulin administration may impair for a period of at least 6 weeks and up to 3 months the efficacy of live attenuated virus vaccines such as measles, rubella, mumps, and varicella. After administration of this medicinal product, an interval of 3 months should elapse before vaccination with live attenuated virus vaccines. In the case of measles, this impairment may persist for up to 1 year. Therefore, patients receiving measles vaccine should have their antibody status checked.
Loop diuretics: Avoidance of concomitant use of loop diuretics.
Paediatric population: Although limited data is available, it is expected that the same interactions may occur in the paediatric population.
Special precautions for disposal and other handling: Privigen comes as a ready-to-use solution in single-use vials. The product should be brought to room temperature (25°C) before use. A vented infusion line should be used for the administration of Privigen. Flushing of the infusion tubes with physiological saline or 5% glucose solution is permitted. Always pierce the stopper at its centre, within the marked area.
The solution should be clear or slightly opalescent and colourless or pale yellow. Solutions that are cloudy or have deposits should not be used.
If dilution is desired, 5% glucose solution should be used. For obtaining an immunoglobulin solution of 50 mg/ml (5%), Privigen 100 mg/ml (10%) should be diluted with an equal volume of the 5% glucose solution. Aseptic technique must be strictly observed during the dilution of Privigen.
Any unused medicinal product or waste material should be disposed of in accordance with local requirements.
Incompatibilities: This medicinal product must not be mixed with other medicinal products, diluents, or solvents except those previously mentioned in Special precautions for disposal and other handling.
Do not store above 25°C.
Do not freeze.
Do not use if Privigen has been frozen.
Do not shake.
Keep the vial in the outer carton in order to protect from light.
Shelf life: 3 years.
Stability after first opening: Once the vial has been broached, its contents should be used promptly. Because the solution contains no preservative, Privigen should be infused immediately.
Stability after dilution: If the product is diluted to lower concentrations (see Special precautions for disposal and other handling under Cautions for Usage), immediate use after dilution is recommended. The in-use stability of Privigen after dilution with a 5% glucose solution to a final concentration of 50 mg/ml (5%) has been demonstrated for 10 days at 30°C; however, the microbial contamination aspect was not studied.
J06BA02 - immunoglobulins, normal human, for intravascular adm. ; Belongs to the class of normal human immunoglobulins. Used in passive immunizations.
Privigen soln for infusion 5 g/50 mL
1's



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image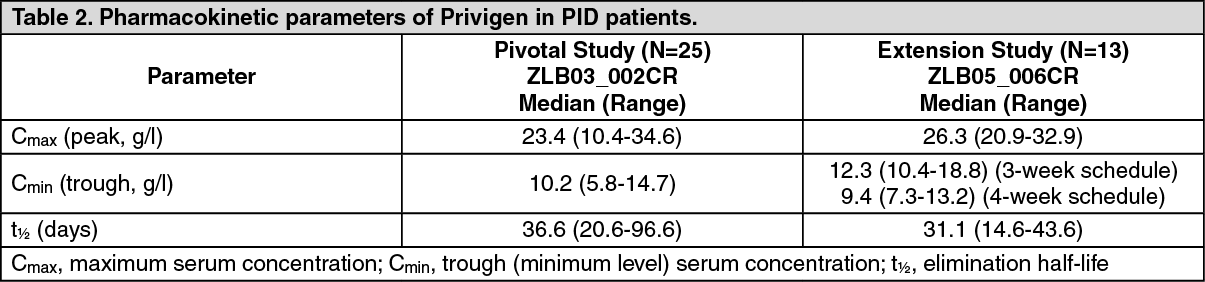 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image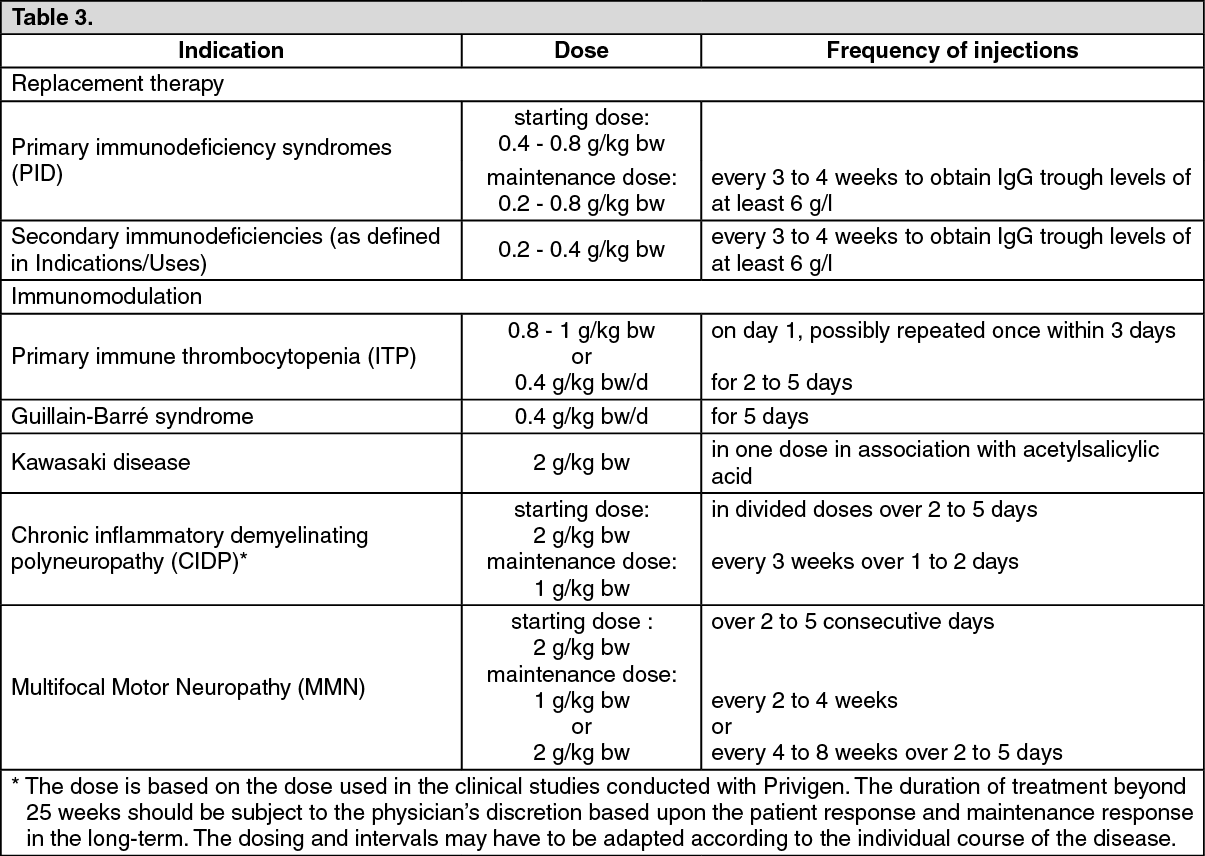 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image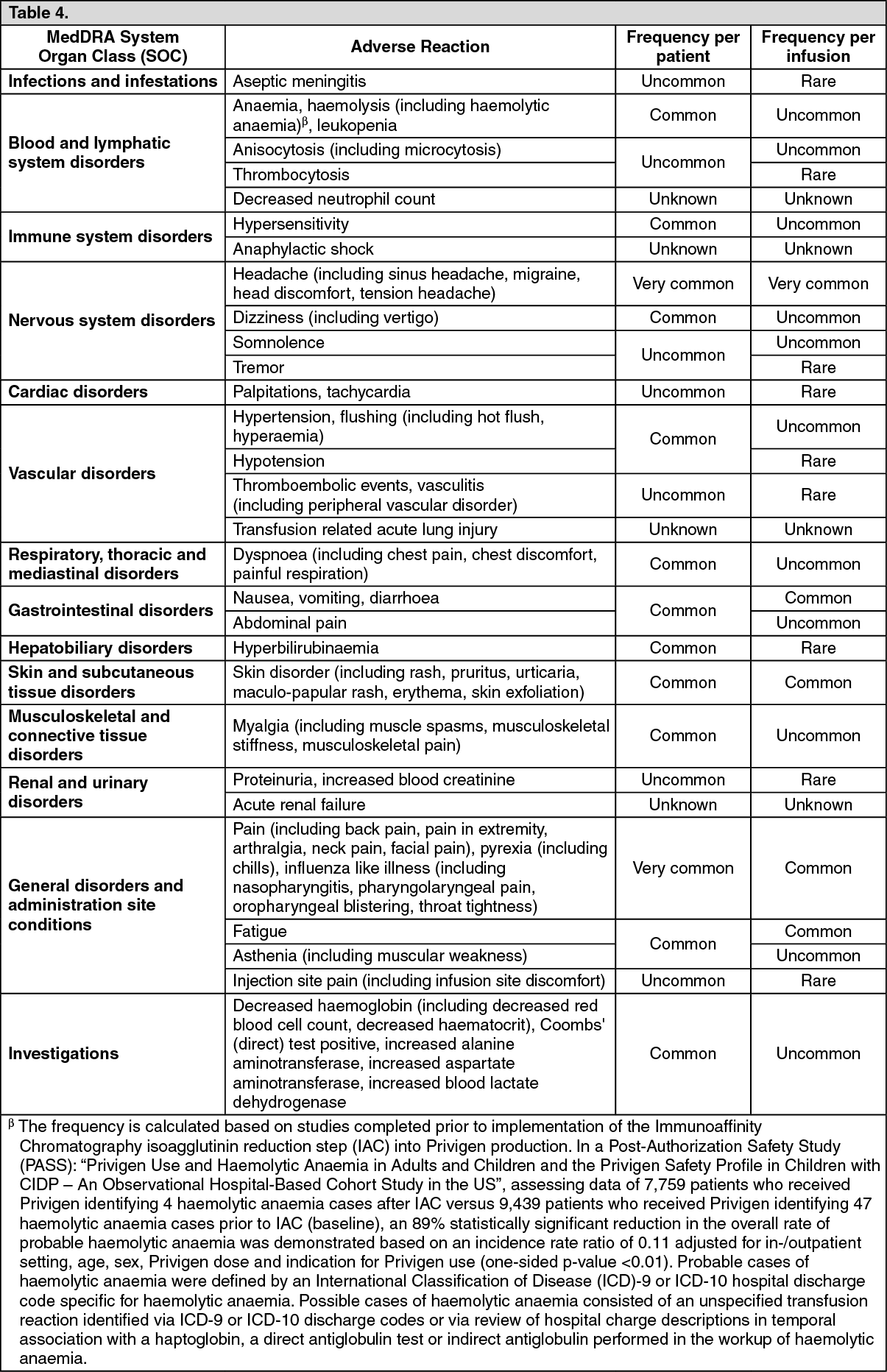 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
