


 Sign Out
Sign Out




Pharmacodynamics: Brexpiprazole has high affinity (expressed as Ki values) for serotonin 5HT1A (0.12 nM), 5HT2A (0.47 nM), 5HT2B (1.88 nM), dopamine D2 (0.3 nM), D3 (1.14 nM), and noradrenergic α1A (3.78 nM), α1B (0.17 nM), α1D (2.60 nM), and α2C (0.59 nM) receptors.
Brexpiprazole exhibits a moderate affinity for dopamine D4 (6.3 nM), serotonin 5-HT7A (9.48 nM), noradrenergic α2A (15 nM), α2B (17 nM) and histamine H1 (19 nM) receptors; and weak affinity for the serotonin 5-HT1B (32 nM) and 5-HT2C (33 nM) receptors (see Detailed Pharmacology as follows).
Brexpiprazole acts as a partial agonist at the 5-HT1A, D2, and D3 receptors and as an antagonist at 5HT2A, 5HT2B, 5HT7, α1A, α1B, α1D, and α2C receptors.
Cardiac Electrophysiology: In a multicenter, randomized, double-blind, placebo- and positive-controlled, parallel group, multiple dose ECG assessment study, subjects with schizophrenia or schizoaffective disorder received treatment with brexpiprazole at a therapeutic dose of 4 mg/day or a supratherapeutic dose of 12 mg/day for 11 days. On day 11, the maximum placebo-adjusted mean change from baseline in the QTcI interval was 8.3 ms (90% CI 3.7, 12.9) at 6 h post-dosing in the brexpiprazole 4 mg/day group (N=62) and 3.1 ms (90% CI -1.7, 8.0) at 4 h post-dosing in the brexpiprazole 12 mg/day group (N=53). No exposure-response relationship was apparent.
Sub-group analyses suggested that the QTc prolongation was larger in female subjects than in males. In the brexpiprazole 4 mg/day group, the maximum placebo-adjusted mean change from baseline in the QTcI interval was 5.2 ms (90% CI 1.5, 8.9) in males (N=48) and 15.0 ms (90% CI 7.7, 22.3) in females (N=14) at 6 h post-dosing. In the brexpiprazole 12 mg/day group, the maximum placebo-adjusted mean change from baseline in the QTcI interval was 2.9 ms (90% CI -1.2, 6.9) in males (N=40) at 12 h post-dosing and 10.4 ms (90% CI 2.7, 18.2) in females (N=13) at 24 h post-dosing. Limitations of the gender sub-group analyses included diminished statistical power.
The brexpiprazole 4 mg/day treatment had no effect on heart rate; however, the brexpiprazole 12 mg/day treatment was associated with an increase in heart rate, with a maximum mean difference from placebo of 4.8 bpm (90% CI 1.9, 7.7) at 2 h.
Clinical Trials: Trial Design and Study Demographics: Schizophrenia: The efficacy of REXULTI in the treatment of adults with schizophrenia was demonstrated in two 6-week, randomized, double-blind, placebo-controlled fixed-dose clinical trials and one longer-term randomized-withdrawal trial in subjects who met DSM-IV-TR criteria for schizophrenia and were experiencing an acute exacerbation of psychotic symptoms. The efficacy was also evaluated in a 6-week, randomized, double-blind, placebo-controlled and active-reference flexible-dose clinical trial.
In two fixed-dose trials, Trial 231 (hereafter "Trial 1") and Trial 230 (hereafter "Trial 2"), subjects were randomized to REXULTI 2 or 4 mg once per day or placebo. Subjects in the REXULTI groups initiated treatment at 1 mg once daily on Days 1 to 4. The REXULTI dosage was increased to 2 mg on Days 5 to 7. The dosage was then either maintained at 2 mg once daily or increased to 4 mg once daily, depending on treatment assignment, for the 5 remaining weeks. The primary efficacy endpoint of both trials was the change from baseline to Week 6 in the Positive and Negative Syndrome Scale (PANSS) total score. The PANSS is a 30-item scale that measures positive symptoms of schizophrenia (7 items), negative symptoms of schizophrenia (7 items), and general psychopathology (16 items), each rated on a scale of 1 (absent) to 7 (extreme); the total PANSS scores range from 30 (best) to 210 (worst). The key secondary endpoint of both trials was the change from baseline to Week 6 in Clinical Global Impression Severity of Illness Scale (CGI-S) total score, a validated clinician-related scale that measures the subject's current illness state and overall clinical state on a 1 (normal, not at all ill) to 7-point (extremely ill) scale.
The longer term Trial 3 was a randomized-withdrawal, double-blind, placebo-controlled trial to assess the efficacy of REXULTI (1 - 4 mg/day) in adults with schizophrenia experiencing an exacerbation of psychotic symptoms at study entry, who met criteria for stability for at least 12 weeks during single-blind treatment with REXULTI (flexible doses 1- 4 mg/day), and were then randomized to continue on their REXULTI dose or to switch to placebo, for up to 52 weeks. The primary endpoint was the time to exacerbation of psychotic symptoms/impending relapse; the key secondary endpoint was the percentage of subjects with exacerbation of psychotic symptoms/impending relapse.
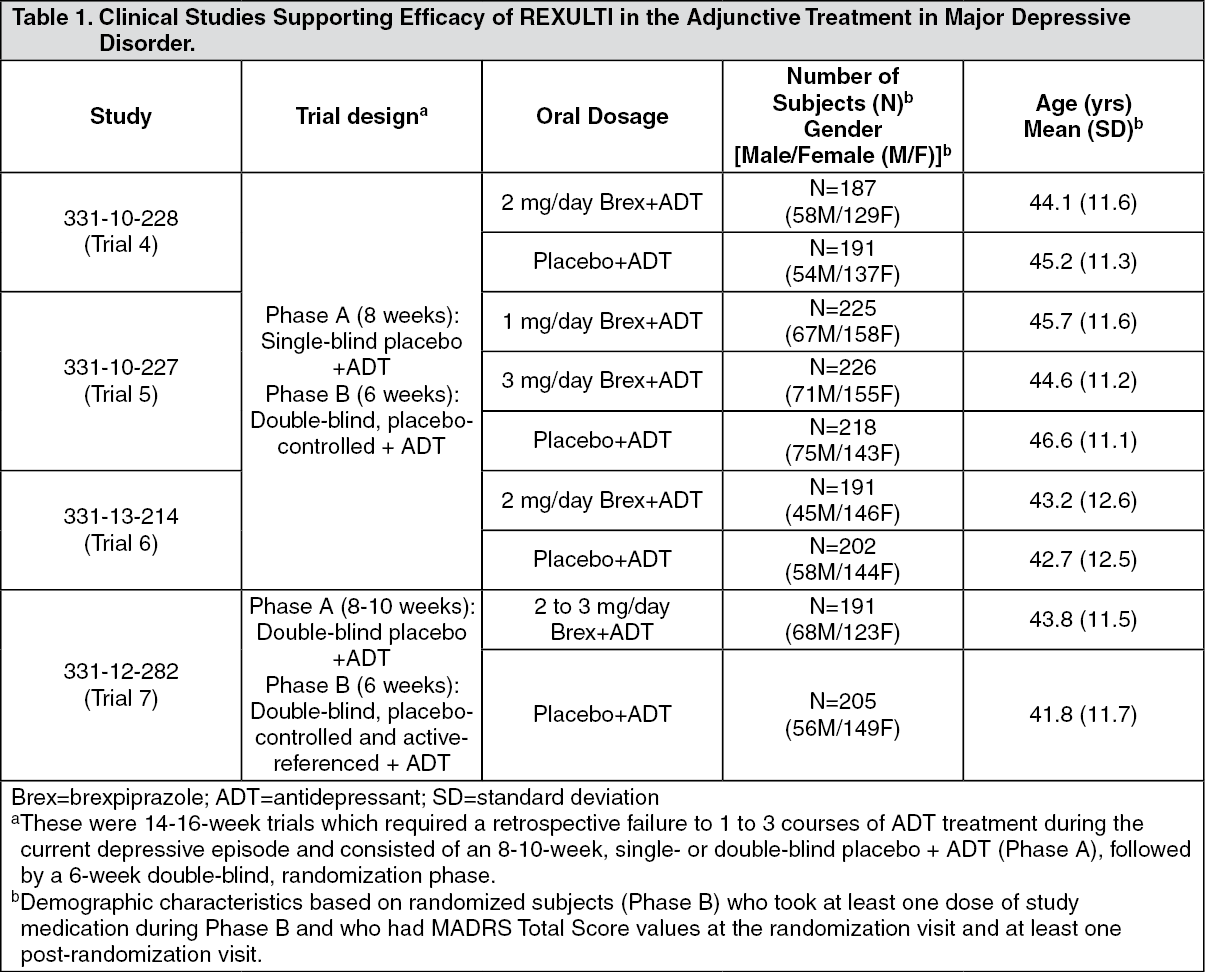
Adjunctive Treatment in Major Depressive Disorder (MDD): The efficacy of REXULTI, as an adjunctive treatment to antidepressant therapy for major depressive disorder (MDD), was evaluated in four phase 3, 6-week, double-blind, placebo-controlled trials: three fixed-dose trials (331-10-228, 331-10-227, 331-13-214) and one flexible-dose trial with an active reference (331-12-282). These trials are referred to as Trials 4, 5, 6 and 7, respectively, in Table 1.
The adult patients in these trials fulfilled the DSM-IV-TR criteria for MDD, with or without symptoms of anxiety, and demonstrated an inadequate response (patient reported) to 1-3 prior antidepressant therapy(ies) in the current episode and an inadequate response during the 8-10 weeks of prospective antidepressant treatment (escitalopram, fluoxetine, paroxetine controlled-release, sertraline, duloxetine or venlafaxine extended-release) during the trials. Inadequate response to prospective antidepressant treatment in Studies 4 and 5 was initially defined as < 50% improvement from baseline on the Hamilton Depression scale (HAMD-17), a HAMD-17 score > 14, and a Clinical Global Impression (CGI-I) ≥ 3 at Week 8. To ensure that randomized patients had an inadequate response throughout the prospective antidepressant treatment phase, this definition was amended during Studies 4 and 5 to the following: < 50% improvement from baseline on the HAMD-17 and a HAMD-17 score > 14 at Week 8; and, CGI-I ≥ 3 and < 50% improvement from baseline on the Montgomery-Asberg Depression Rating Scale (MADRS) Total Score at Weeks 2, 4, 6 and 8 (and Week 10, as applicable). This definition of inadequate response to prospective antidepressant treatment was also applied in Study 6. With the exception of approximately 6% of patients in Studies 4 and 5, all patients who were randomized in the short-term clinical trials 4, 5, and 6 fulfilled the revised definition of inadequate response to prospective antidepressant treatment. In Study 7, as the HAMD-17 was not administered, a MADRS total score ≥18 at the end of prospective treatment was used in lieu of HAMD-17 score ≥14.
Patients remained on the same antidepressant treatment throughout the entire duration of each study. All patients randomized to REXULTI in the fixed-dose studies (Studies 4, 5 and 6) initiated treatment at 0.5 mg/day during Week 1. The REXULTI dose was increased to 1 mg/day during Week 2 in all dose groups and, based on the assigned treatment, the dose was either maintained at 1 mg/day or increased to 3 mg/day (Study 5) or increased to 2 mg/day (Studies 4 and 6), from Week 3 onwards. Dosages were maintained at the assigned doses for the 4 remaining weeks. In the flexible-dose study (Study 7), patients randomized to REXULTI initiated treatment at 1 mg/day during Week 1, and the dose was increased to the target dose of 2 mg/day during Week 2. Patients remained at 2 mg/day in Study 7 unless there was a decision to increase the dose to 3 mg/day.
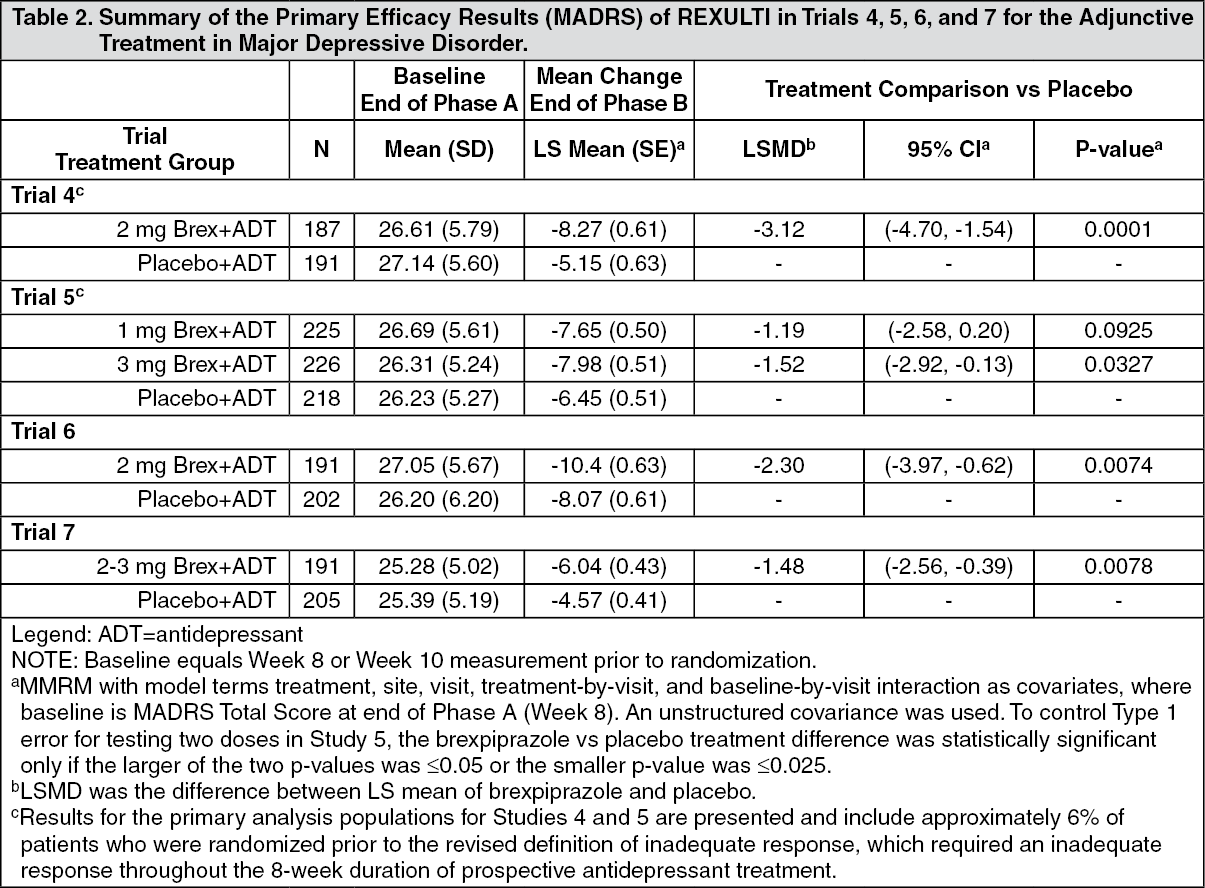
The primary efficacy endpoint in all studies was mean change from baseline (randomization) to Week 6 on the Montgomery Asberg Depression Rating Scale (MADRS) Total Score, a 10-item clinician-rated scale that assesses the degree of depressive symptomatology (apparent sadness, reported sadness, inner tension, reduced sleep, reduced appetite, concentration difficulties, lassitude, inability to feel, pessimistic thoughts, and suicidal thoughts). Each item is scored from 0 (normal/symptom not present) to 6 (most severe symptoms) and the range for the total score is 0 to 60.
The key secondary instrument was the Sheehan Disability Scale (SDS), a 3-item self-rated instrument used to assess three domains of functioning (work/school, social life, and family life) with each item scored from 0 (no disruption at all) to 10 (extreme disruption). (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageStudy Results: Schizophrenia: In Trial 1, REXULTI was superior to placebo (N=178) at both 2 mg/day (n=180) and 4 mg/day (N=178) doses for the primary endpoint (PANSS total score) and key secondary endpoint (CGI-S total score).
In Trial 2, REXULTI at 4 mg/day group (N=181) was superior to placebo (N=180) for the primary endpoint (PANSS total score), but not at the 2 mg/day dose (N=179).
Examination of population subgroups based on age, gender and race did not suggest differential responsiveness.
In the 6-week flexible-dose study (Study 14644A), REXULTI at doses between 2 and 4 mg/day (N=150) was not superior to placebo (N=159) for the primary endpoint, the mean changes in PANSS total score at Week 6; however, the active reference (N=150) confirmed the assay sensitivity of the study.
In the longer-term Trial 3, pre-specified interim analysis, conducted after 50% of the events planned in the calculation of power, demonstrated a statistically significantly longer time to relapse in subjects randomized to the REXULTI group compared to placebo-treated subjects and the trial was subsequently terminated early because of demonstrated efficacy. The final analysis demonstrated a statistically significantly longer time to relapse in subjects randomized to the REXULTI group (N=96) compared to placebo-treated subjects (N=104). Time to impending relapse was statistically significantly delayed with REXULTI compared with placebo in both the interim and final analyses (p = 0.0008 and p < 0.0001, respectively; log-rank test). For the final analysis, the hazard ratio from the Cox proportional hazard model for the placebo to REXULTI comparison was 3.420 (95% CI: 1.825, 6.411); thus, subjects in the placebo group had a 3.4-fold greater risk of experiencing impending relapse than the subjects in the REXULTI group.
The key secondary endpoint of Trial 3, the proportion of subjects who met the criteria for impending relapse, was statistically significantly lower in REXULTI-treated subjects compared with placebo group (13.5% vs. 38.5%, p<0.0001).
Adjunctive Treatment in Major Depressive Disorder (MDD).
For the randomized patients in Trials 4-7, the mean duration of the current major depressive episode ranged between approximately 12 and 18 months and the majority of patients (approximately 79% - 84%) reported an inadequate response to one prior antidepressant treatment, before receiving 8-10 weeks of prospective antidepressant treatment during the trials. Following 8-10 weeks of prospective antidepressant treatment, the mean MADRS Total Score at randomization ranged between 25 and 27. Mean SDS score at randomization was between 5.6 and 6.3.
In Trials 4, 6 and 7 there was greater improvement in the mean MADRS Total Score with REXULTI (2 mg/day or 2-3 mg/day) + ADT compared to placebo + ADT (p < 0.05). No additional benefit was demonstrated at doses greater than 2 mg/day (Table 2). In Study 7 the majority of patients treated with REXULTI received 2 mg/day and the mean daily REXULTI dose at endpoint was 2.2 mg/day. (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn Trial 4, the mean SDS score showed greater improvement with REXULTI (2 mg/day) + ADT than with placebo + ADT (p<0.05).
Detailed Pharmacology: Brexpiprazole has a broad receptor binding profile with high affinity (Ki < 5 nM) for multiple monoaminergic receptors including serotonin 5-HT1A, 5-HT2A, 5-HT2B, dopamine D2, D3, and noradrenergic α1A, α1B, α1D, and α2C receptors. Brexpiprazole exhibits a moderate affinity for dopamine D4, serotonin 5-HT7A, noradrenergic α2A, α2B and histamine H1 receptors; and weak affinity for the serotonin 5-HT1B and 5-HT2C receptors. Although affinity constants have not been determined, brexpiprazole (at 10 μM) showed occupancy at the muscarinic M1 receptor (64%), dopamine transporter (90%) and serotonin transporter (65%).
Brexpiprazole acts as a partial agonist at the 5-HT1A, D2, and D3 receptors and as an antagonist at 5-HT2A, 5-HT2B, 5HT7, α1A, α1B, α1D, and α2C receptors. Dose response occupancy and brain/plasma exposure relationship were determined in vivo or ex vivo for D2/D3, 5-HT2A, 5-HT1A, and 5-HT7 receptors as well as the serotonin transporter in preclinical studies. These results are consistent with the relative binding affinities and indicate that brexpiprazole has activity at several targets in the central nervous system at therapeutic plasma exposures.
Brexpiprazole was shown in vitro to inhibit both norepinephrine and serotonin uptake into synaptosome preparations from rat brain tissue. Brexpiprazole also inhibited monoamine oxidase B (MAO-B) enzyme activity in rat liver extracts.
Central Nervous System Safety Pharmacology: In safety pharmacology studies, brexpiprazole had a depression effect on the CNS that was related to the exaggerated pharmacological effect of the compound. Brexpiprazole caused a decreased body temperature in repeat-dose toxicity studies at doses ≥30 mg/kg in rats, monkeys and dogs (see Toxicology: Repeat-Dose Toxicity and Juvenile Repeat-Dose Toxicity as follows).
Cardiovascular Safety Pharmacology: Significant decrease in blood pressure and prolongation of QT interval and QTc were noted in the conscious telemetry dog trial, and on Day 1 of administration at doses ≥3 mg/kg in the repeat-dose toxicity studies with monkeys and in the juvenile toxicity study with dogs (15- and 24-fold the MRHD on a mg/m2 basis, respectively). In conscious telemetry dogs (N=4), brexpiprazole was administered at sequential oral doses of 0 (vehicle), 1, 3, 10, and 30 mg/kg at intervals of 7-8 days. Brexpiprazole at 10 mg/kg and 30 mg/kg caused statistically significant increases in the QTc interval and the QRS duration compared to the vehicle control group.
In anesthetised dogs (N=4) under phenylephrine-induced hypertensive state, intravenous infusions of brexpiprazole were associated with statistically significant decreases in systolic and diastolic blood pressure at 0.3 mg/kg and 3 mg/kg and decrease in heart rate at 3 mg/kg. The effect of brexpiprazole on blood pressure may be due to a blockade of α1-adrenoceptors in peripheral blood vessels, which is consistent with the pharmacological profile for this compound.
In Chinese hamster ovary cells CHO-K1 expressing the alpha subunit of the human IKr potassium channel, brexpiprazole caused a statistically significant and concentration-dependent suppression of hERG currents over a 0.01 to 1 μM concentration range, with a IC50 of 0.117 μM (51 ng/mL).
Pharmacokinetics: Absorption: After single dose administration of REXULTI tablets, the peak plasma brexpiprazole concentrations occurred within 4 hours after administration; and the absolute oral bioavailability was 95%. Brexpiprazole steady-state concentrations were attained within 10-12 days of dosing.
REXULTI can be administered with or without food. Administration of a 4-mg REXULTI tablet with a standard high fat meal did not significantly affect the Cmax or AUC of brexpiprazole. After single and multiple once daily dose administration, brexpiprazole exposure (Cmax and AUC) increased in proportion to the dose administered. In vitro studies of brexpiprazole did not indicate that brexpiprazole is a substrate of efflux transporters such as MDRI (P-gp) and BCRP.
Distribution: The volume of distribution of brexpiprazole following intravenous administration is high (1.56±0.42 L/kg), indicating extravascular distribution. Brexpiprazole is highly protein bound in plasma (greater than 99%) to serum albumin and α1-acid glycoprotein, and its protein binding is not affected by renal or hepatic impairment. Based on results of in vitro studies, brexpiprazole protein binding is not affected by warfarin, diazepam, or digitoxin.
Metabolism: Based on in vitro metabolism studies of brexpiprazole using recombinant human cytochrome P450 (CYP1A1, 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, and 3A4), the metabolism of brexpiprazole was shown to be mainly mediated by CYP3A4 and CYP2D6. Based on in vitro data, brexpiprazole showed little to no inhibition of CYP450 isozymes.
In vivo brexpiprazole is metabolized primarily by CYP3A4 and CYP2D6 enzymes. After single- and multiple-dose administrations, brexpiprazole and its major metabolite, DM-3411, were the predominant drug moieties in the systemic circulation. At steady-state, DM-3411 represented 23% to 48% of brexpiprazole exposure (AUC) in plasma. DM-3411 is considered not to contribute to the therapeutic effects of brexpiprazole.
Excretion: Following a single oral dose of [14C]-labeled brexpiprazole, approximately 25% and 46% of the administered radioactivity was recovered in the urine and feces, respectively. Less than 1% of unchanged brexpiprazole was excreted in the urine and approximately 14% of the oral dose was recovered unchanged in the feces. Apparent oral clearance of brexpiprazole oral tablet after once daily administration is 19.8 (±11.4) mL/h/kg. After multiple once daily administration of brexpiprazole, the terminal elimination half-life of brexpiprazole and its major metabolite, DM-3411, is 91.4 hours and 85.7 hours, respectively.
Special Populations and Conditions: Pediatrics: The safety and efficacy of REXULTI in patients under the age of 18 years have not been established.
The pharmacokinetics, safety and tolerability of brexpiprazole 0.5 - 4 mg per day oral doses were assessed in 43 adolescent subjects (aged 13 to 17 years, weight range 43.4 - 116.2 kg) with a diagnosis of schizophrenia, bipolar disorder, or other related psychiatric disorders in an open-label, dose-escalation trial (Trial 8). The brexpiprazole exposure, in terms of AUC and Cmax, seemed slightly higher and apparent clearance seemed slightly lower in adolescent subjects compared with adult subjects.
The pharmacokinetics, safety and tolerability of brexpiprazole single oral doses of 0.75 and 1.5 mg in 12 subjects 6 to < 10 years old (weight range 20.1 - 40.0 kg) and single oral doses of 1.5 and 3 mg in 12 subjects 10 to < 13 years old (weight range 28.0 - 61.0 kg) with a diagnosis of CNS disorders were assessed in a sequential cohort, nonrandomized crossover trial (Trial 9). Children 6 to < 10 years old appeared to have slightly higher brexpiprazole exposure and lower brexpiprazole apparent clearance as compared to children 10 to < 13 years old.
The pharmacokinetic profile in pediatric patients and the comparison with adults should be considered preliminary. REXULTI is not indicated for use in patients below the age of 18 (see Indications/Uses, and Use in Children under Precautions).
Geriatrics: Clinical studies of the efficacy of REXULTI did not include a meaningful number of subjects aged 65 or older to determine whether they respond differently from younger subjects. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, and cardiac function, concomitant diseases, and other drug therapy.
Antipsychotic drugs increase the risk of death in elderly patients with dementia-related psychosis. REXULTI is not approved for the treatment of patients with dementia-related psychosis (see Increased Mortality in Elderly Patients with Dementia under Warnings).
CYP2D6 poor metabolism status: Based on the results of the population PK analysis CYP2D6 poor metabolizer subjects exhibited 47% higher exposure (AUCτ) to brexpiprazole compared with CYP2D6 extensive metabolizer subjects (see Dosing Considerations: CYP isozymes under Dosage & Administration).
Age/Sex: After single dose administration of brexpiprazole (2 mg), elderly subjects (older than 65 years old) exhibited similar brexpiprazole systemic exposure (Cmax and AUC) in comparison to the adult subjects (18-45 years old) and female subjects exhibited approximately 40-50% higher brexpiprazole systemic exposure (Cmax and AUC) in comparison to the male subjects. Population pharmacokinetic evaluation identified age and female sex as statistically significant covariates affecting brexpiprazole PK while the effects were not considered clinically relevant. No dosage adjustment is required in subjects based on age or sex (see Use in the Elderly: Geriatrics (> 65 years of age) under Precautions).
Ethnic Origin: Although no specific pharmacokinetic study was conducted to investigate the effects of race on the disposition of brexpiprazole, population pharmacokinetic evaluation revealed no evidence of clinically significant race-related differences in the pharmacokinetics of brexpiprazole. No dosage adjustment is required in patients based on race.
Hepatic Insufficiency: In subjects with varying degrees of hepatic impairment (Child-Pugh Classes A, B, and C; N=22), the AUC of oral brexpiprazole (2 mg single dose), compared to matched healthy subjects, increased 24% in mild hepatic impairment, increased 60% in moderate hepatic impairment, and 8% in severe hepatic impairment. Specific dosing considerations are recommended for patients with moderate to severe hepatic impairment (see Dosing Considerations: Hepatic Impairment under Dosage & Administration).
Renal Insufficiency: In subjects with severe renal impairment (CLcr <30 mL/min; N=10), AUC of oral brexpiprazole (2 mg single dose) compared to matched healthy subjects was increased by 68% while its Cmax was not changed. Specific dosing considerations are recommended for patients with moderate, severe or end stage renal impairment (see Dosing Considerations: Renal Impairment under Dosage & Administration).
Toxicology: Non-Clinical Toxicology: Acute Toxicity: In single-dose oral (gavage) toxicity studies, the minimum oral lethal dose was >1000 and 300 mg/kg, respectively for male and female Sprague Dawley (SD) rats, and > 100 mg/kg for both male and female cynomolgus monkeys. At doses of 1000 and 300 mg/kg, clinical signs observed in male and female rats included hypoactivity, closed eyes or incomplete eyelid closure, fixed stare, lacrimation, abnormal posture, and hypothermia. In monkey, clinical signs included drowsiness, partially closed eyes, crouching or prone positions, tremors of the limbs, decrease in movement, and decrease in body temperature.
Repeat-Dose Toxicity: In a repeat-dose toxicity study conducted in rats at oral doses of 0, 3, 10, 30 and 100 mg/kg/day for 26 weeks duration, the no observed adverse effect level (NOAEL) was 3 mg/kg (7-fold MRHD on a mg/m2 basis). Clinical signs observed at 30 and 100 mg/kg included CNS depression, hypoactivity, hypothermia, gynecomastia, galactorrhea, and increases in blood levels of aspartate aminotransferase and gamma globulin, as well as decrease in body weight and food consumption. Female rats increased in body weight at 3 mg/kg compared to the control group. Major histopathology finding corresponded to atrophy of the uterus, thymus and pituitary glands, and enlargement of adrenal glands at doses ≥10 mg/kg.
In repeat-dose toxicity studies conducted in Cynomolgus monkeys at oral doses of 0, 1, 3 and 30 mg/kg/day for 13 and 39 weeks duration, the NOAEL was 1 mg/kg (5-fold MRHD) for both sexes. At doses ≥3 mg/kg, animals exhibited CNS depression, decrease in blood pressure, decreases in leukocytes and reticulocytes, as well as increases in blood cholesterol and phospholipids. Major histopathology finding corresponded to death-related gastrointestinal bleeding and ulcers at 30 mg/kg.
Brexpiprazole caused a decreased body temperature in repeat-dose toxicity studies at doses ≥30 mg/kg in rats, monkeys and dogs.
Significant decrease in blood pressure and prolongation of QT interval and QTc were noted in the conscious telemetry dog trial, and on Day 1 of administration at doses ≥ 3 mg/kg in the repeat-dose toxicity studies with monkeys and in the juvenile toxicity study with dogs (15- and 24-fold the MRHD on a mg/m2 basis, respectively). In conscious telemetry dogs (N=4), brexpiprazole was administered at sequential oral doses of 0 (vehicle), 1, 3, 10, and 30 mg/kg at intervals of 7-8 days. Brexpiprazole at 10 mg/kg and 30 mg/kg caused statistically significant increases in the QTc interval and the QRS duration compared to the vehicle control group.
Juvenile Repeat-Dose Toxicity: In repeat-dose toxicity study conducted in juvenile rats at oral doses of 0, 3, 10 and 20 mg/kg/day for 8 weeks duration, the NOAEL was <3 mg/kg (7-fold MRHD on a mg/m2 basis) in both sexes. At doses ≥10mg/kg, animals exhibited CNS depression, hypoactivity, as well as increases in blood globulins and phospholipids. Decrease in body weight, pubertal delays and gynecomastia were also noted at the end of the administration period compared with the control group. Female rats increased in body weight at 3 mg/kg. Major histopathology finding corresponded to atrophy of the pituitary glands, liver and kidney at doses ≥10 mg/kg. Following 2 weeks untreated recovery period, differences in fertility and reproductive performance between treatment groups were unremarkable.
In repeat-dose toxicity study conducted in juvenile Beagle dogs at oral doses of 0, 1, 3 and 30 mg/kg/day for 26 weeks duration, the NOAEL was <3 mg/kg (24-fold MRHD) in both sexes. At 30 mg/kg, animals exhibited CNS depression, hypoactivity, decreased respiration rates, lower blood pressure and decrease in reticulocytes, as well as, increases in blood cholesterol and phospholipids. Major histopathology finding corresponded to enlargement of adrenal glands and liver at 30 mg/kg. Toxicology findings were unremarkable after 8 weeks untreated recovery period, except for male pubertal delays noted in the 30 mg/kg group.
Carcinogenesis: The lifetime carcinogenic potential of brexpiprazole was evaluated in two-year studies in mice and rats. In mice, brexpiprazole was administered orally (gavage) at doses of 0.75, 2 and 5 mg/kg/day (1- to 6-fold MRHD on a mg/m2 basis). There was no increase in the incidence of tumors in males at any dose group. In female mice, there was an increased incidence of mammary gland adenocarcinoma and adenosquamous carcinoma, and pars distalis adenoma of the pituitary gland at all doses. In rats, brexpiprazole was administered orally (gavage) at doses of 1, 3 and 10 mg/kg/day in males or 3, 10 and 30 mg/kg/day in females (for males 2- to 24-fold and for females 7- to 73-fold the MRHD). Long-term administration of brexpiprazole to rats did not induce neoplastic lesions.
Proliferative and/or neoplastic changes in the mammary and pituitary glands of rodents have been observed following chronic administration of antipsychotic drugs and are considered to be prolactin-mediated. The potential for increasing serum prolactin level of brexpiprazole was shown in both sexes in mice and rats. The relevance for human risk of the findings of prolactin-mediated endocrine tumors in rodents is unknown.
Mutagenesis: Brexpiprazole was not mutagenic when tested in the in vitro bacterial reverse-mutation assay (Ames test), in vivo in the micronucleus assay in rats, and the unscheduled DNA synthesis assay in rats. Brexpiprazole was mutagenic and clastogenic but occurred only at doses that induced cytotoxicity (20-30 μg/mL) in the in vitro forward gene mutation assay in mouse lymphoma cells and in the in vitro chromosomal aberration assay in Chinese hamster ovary cells. Based on a weight of evidence, brexpiprazole is not considered to present a genotoxic risk to humans at therapeutic doses and exposures.
Impairment of Fertility: In female rats treated with brexpiprazole at oral doses of 0, 0.3, 3 or 30 mg/kg/day prior to mating with untreated males and continuing through conception and implantation, the NOAEL in terms of reproductive performance and fertility was 0.3 mg/kg/day (0.7-fold MRHD). Prolonged diestrus and decreased fertility were observed at 3 mg/kg/day. At 30 mg/kg/day a prolongation of the mating phase was observed and significantly increased preimplantation losses were seen.
In male rats treated with brexpiprazole at oral doses of 0, 3, 10 or 100 mg/kg/day for 63 days prior to mating and during copulation (with untreated females), the NOAEL in terms of male fertility and toxicological effects was 10 mg/kg/day (24-fold MRHD).
Reproductive Toxicity: In a prenatal and postnatal developmental study in rats, pregnant dams receiving brexpiprazole at oral doses of 0, 3, 10 and 30 mg/kg/day from implantation until weaning of offspring, the NOAEL for maternal toxicity and newborn viability was 3 mg/kg/day (7-fold MRHD). Increase in the number of stillbirth and death in pups during lactation were observed at 10 and 30 mg/kg. Changes observed at 30 mg/kg/day included impaired nursing in dams, and low birth weight, impaired viability, suppressed body weight gain, delayed pinna unfolding and decreased number of corpora lutea in the offspring.
In a rabbit embryo-fetal development study, pregnant dams receiving brexpiprazole at oral doses of 0, 10, 30 and 150 mg/kg/day during gestation, the NOAEL for reproductive toxicity was 10 mg/kg/day (49-fold MRHD). At doses ≥30 mg/kg, an increase incidence in renal pelvic dilation and caudal vena cava abnormality was observed in fetuses. At 150 mg/kg/day, decreased body weight, retarded ossification, and increased incidences of visceral and skeletal malformations were observed.