


 Sign Out
Sign Out
Pharmacology: Pharmacodynamics: Mechanism of action: Upadacitinib is a selective and reversible Janus kinase (JAK) inhibitor. JAKs are intracellular enzymes that transmit cytokine or growth factor signals involved in a broad range of cellular processes including inflammatory responses, hematopoiesis, and immune surveillance. The JAK family of enzymes contains four members, JAK1, JAK2, JAK3 and TYK2 which work in pairs to phosphorylate and activate signal transducers and activators of transcription (STATs). This phosphorylation, in turn, modulates gene expression and cellular function. JAK1 is important in inflammatory cytokine signals while JAK2 is important for red blood cell maturation and JAK3 signals play a role in immune surveillance and lymphocyte function.
In human cellular assays, upadacitinib preferentially inhibits signalling by JAK1 or JAK1/3 with functional selectivity over cytokine receptors that signal via pairs of JAK2. Atopic dermatitis is driven by pro-inflammatory cytokines (including IL-4, IL-13, IL-22, TSLP, IL-31 and IFN-γ) that transduce signals via the JAK1 pathway. Inhibiting JAK1 with upadacitinib reduces the signaling of many mediators which drive the signs and symptoms of atopic dermatitis such as eczematous skin lesions and pruritus. Pro-inflammatory cytokines (primarily IL-6, IL-7, IL-15 and IFNγ) transduce signals via the JAK1 pathway and are involved in the pathology of inflammatory bowel diseases. JAK1 inhibition with upadacitinib modulates the signalling of the JAK-dependent cytokines underlying the inflammatory burden and signs and symptoms of inflammatory bowel diseases.
Pharmacodynamic effects: Inhibition of IL-6 induced STAT3 and IL-7 induced STAT5 phosphorylation: In healthy volunteers, the administration of upadacitinib (immediate-release formulation) resulted in a dose- and concentration-dependent inhibition of IL-6 (JAK1/JAK2)-induced STAT3 and IL-7 (JAK1/JAK3)-induced STAT5 phosphorylation in whole blood. The maximal inhibition was observed 1 hour after dosing which returned to near baseline by the end of dosing interval.
Lymphocytes: In patients with rheumatoid arthritis, treatment with upadacitinib was associated with a small, transient increase in mean ALC from baseline up to week 36 which gradually returned to at or near baseline levels with continued treatment.
hsCRP: In patients with rheumatoid arthritis, treatment with upadacitinib was associated with decreases from baseline in mean hsCRP levels as early as week 1 which were maintained with continued treatment.
Vaccine studies: The influence of upadacitinib on the humoral response following administration of adjuvanted recombinant glycoprotein E herpes zoster vaccine was evaluated in 93 patients with rheumatoid arthritis under stable treatment with upadacitinib 15 mg. 98% of patients were on concomitant methotrexate. 49% of patients were on oral corticosteroids at baseline. The primary endpoint was the proportion of patients with a satisfactory humoral response defined as ≥4-fold increase in pre-vaccination concentration of anti-glycoprotein E titer levels at week 16 (4 weeks post-dose 2 vaccination). Vaccination of patients treated with upadacitinib 15 mg resulted in a satisfactory humoral response in 79/90 (88% [95% CI: 81.0, 94.5]) of patients at week 16.
The influence of upadacitinib on the humoral response following the administration of inactivated pneumococcal polysaccharide conjugate vaccine (13-valent, adsorbed) was evaluated in 111 patients with rheumatoid arthritis under stable treatment with upadacitinib 15 mg (n=87) or 30 mg (n=24). 97% of patients (n=108) were on concomitant methotrexate. The primary endpoint was the proportion of patients with satisfactory humoral response defined as ≥2-fold increase in antibody concentration from baseline to week 4 in at least 6 out of the 12 pneumococcal antigens (1, 3, 4, 5, 6B, 7F, 9V, 14, 18C, 19A, 19F, and 23F). Results at week 4 demonstrated a satisfactory humoral response in 67.5% (95% CI: 57.4, 77.5) and 56.5% (95% CI: 36.3, 76.8) of patients treated with upadacitinib 15 mg and 30 mg, respectively.
Clinical efficacy and safety: Rheumatoid arthritis: The efficacy and safety of upadacitinib 15 mg once daily was assessed in five Phase 3 randomised, double-blind, multicentre studies in patients with moderately to severely active rheumatoid arthritis and fulfilling the ACR/EULAR 2010 classification criteria (see Table 1). Patients 18 years of age and older were eligible to participate. The presence of at least 6 tender and 6 swollen joints and evidence of systemic inflammation based on elevation of hsCRP was required at baseline. Four studies included long-term extensions for up to 5 years, and one study (SELECT-COMPARE) included a long-term extension for up to 10 years.
The primary analysis for each of these studies included all randomised subjects who received at least 1 dose of upadacitinib or placebo, and non-responder imputation was used for categorical endpoints.
Across the Phase 3 studies, the efficacy seen with upadacitinib 15 mg QD was generally similar to that observed with upadacitinib 30 mg QD. (See Table 1.)
 Click on icon to see table/diagram/image
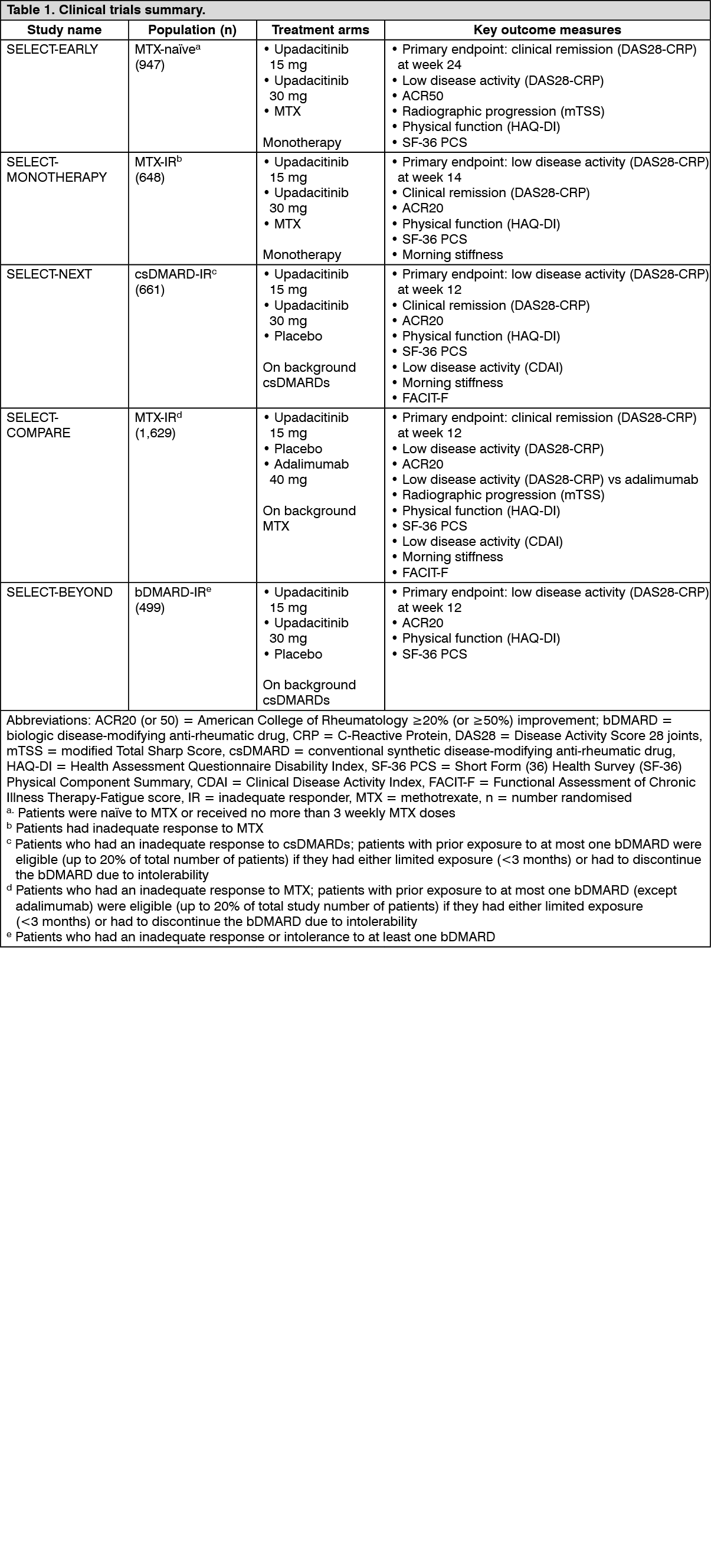
Click on icon to see table/diagram/imageClinical response: Remission and low disease activity: In the studies, a significantly higher proportion of patients treated with upadacitinib 15 mg achieved low disease activity (DAS28-CRP ≤3.2) and clinical remission (DAS28-CRP <2.6) compared to placebo, MTX, or adalimumab (Table 2). Compared to adalimumab, significantly higher rates of low disease activity were achieved at week 12 in SELECT-COMPARE. Overall, both low disease activity and clinical remission rates were consistent across patient populations, with or without MTX. At 3 years, 297/651 (45.6%) and 111/327 (33.9%) patients remained on originally randomised treatment of upadacitinib 15 mg or adalimumab, respectively, in SELECT-COMPARE, and 216/317 (68.1%) and 149/315 (47.3%) patients remained on originally randomised treatment of upadacitinib 15 mg or MTX monotherapy, respectively, in SELECT-EARLY. Among the patients who remained on their originally allocated treatment, low disease activity and clinical remission were maintained through 3 years.
ACR response: In all studies, more patients treated with upadacitinib 15 mg achieved ACR20, ACR50, and ACR70 responses at 12 weeks compared to placebo, MTX, or adalimumab (Table 2). Time to onset of efficacy was rapid across measures with greater responses seen as early as week 1 for ACR20. Durable response rates were observed (with or without MTX), with ACR20/50/70 responses maintained through 3 years among the patients who remained on their originally allocated treatment.
Treatment with upadacitinib 15 mg, alone or in combination with csDMARDs, resulted in improvements in individual ACR components, including tender and swollen joint counts, patient and physician global assessments, HAQ-DI, pain assessment and hsCRP. (See Table 2.)
 Click on icon to see table/diagram/image
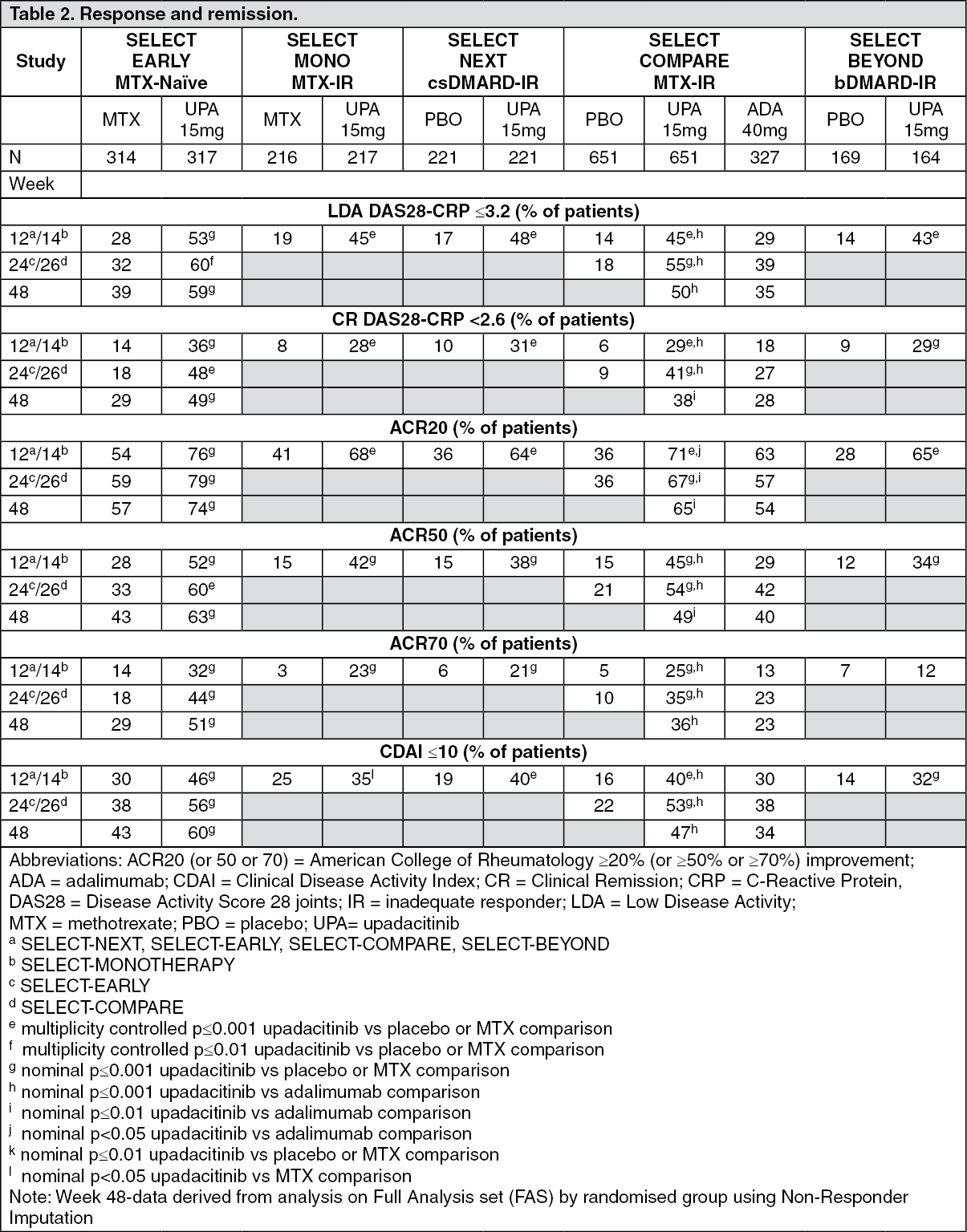
Click on icon to see table/diagram/imageRadiographic response: Inhibition of progression of structural joint damage was assessed using the modified Total Sharp Score (mTSS) and its components, the erosion score and joint space narrowing score, at weeks 24/26 and week 48 in SELECT-EARLY and SELECT-COMPARE.
Treatment with upadacitinib 15 mg resulted in significantly greater inhibition of the progression of structural joint damage compared to placebo in combination with MTX in SELECT-COMPARE and as monotherapy compared to MTX in SELECT-EARLY (Table 3). Analyses of erosion and joint space narrowing scores were consistent with the overall scores. The proportion of patients with no radiographic progression (mTSS change ≤0) was significantly higher with upadacitinib 15 mg in both studies. Inhibition of progression of structural joint damage was maintained through week 96 in both studies for patients who remained on their originally allocated treatment with upadacitinib 15 mg (based on available results from 327 patients in SELECT-COMPARE and 238 patients in SELECT EARLY). (See Table 3.)
 Click on icon to see table/diagram/image
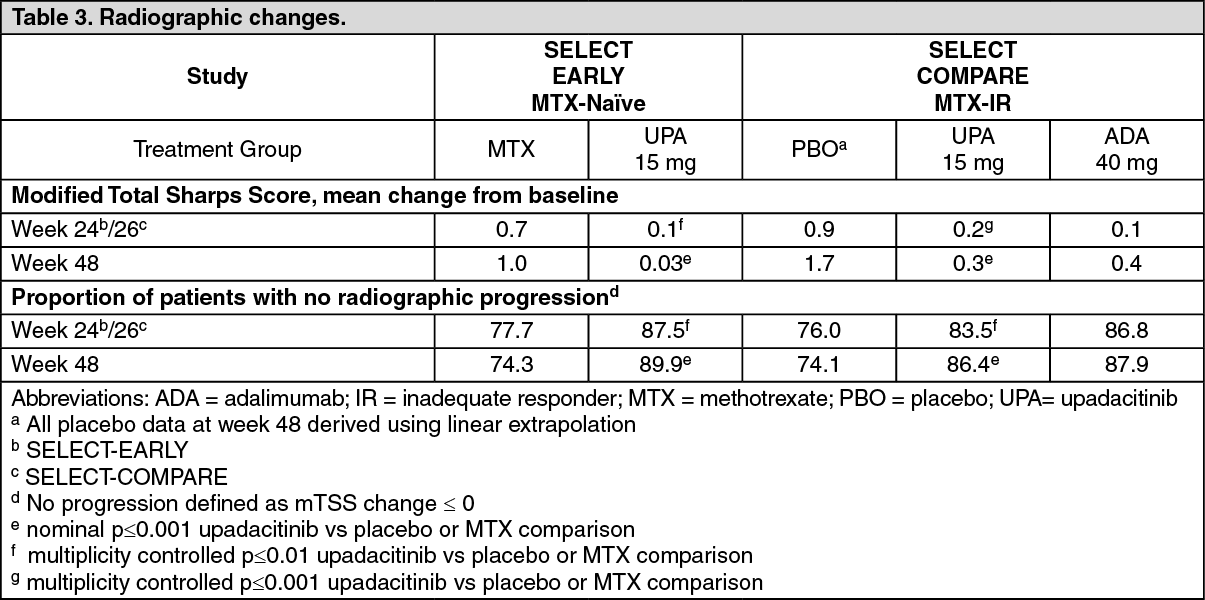
Click on icon to see table/diagram/imagePhysical function response and health-related outcomes: Treatment with upadacitinib 15 mg, alone or in combination with csDMARDs, resulted in a significantly greater improvement in physical function compared to all comparators as measured by HAQ-DI (see Table 4). Improvement in HAQ-DI was maintained through 3 years for patients who remained on their originally allocated treatment with upadacitinib 15 mg based on available results from SELECT-COMPARE and SELECT-EARLY. (See Table 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn the studies SELECT-MONOTHERAPY, SELECT-NEXT, and SELECT-COMPARE, treatment with upadacitinib 15 mg resulted in a significantly greater improvement in the mean duration of morning joint stiffness compared to placebo or MTX.
In the clinical studies, upadacitinib-treated patients reported significant improvements in patient-reported quality of life, as measured by the Short Form (36) Health Survey (SF-36) Physical Component Summary compared to placebo and MTX. Moreover, upadacitinib-treated patients reported significant improvements in fatigue, as measured by the Functional Assessment of Chronic Illness Therapy-Fatigue score (FACIT-F) compared to placebo.
Psoriatic arthritis: The efficacy and safety of upadacitinib 15 mg once daily were assessed in two Phase 3 randomised, double-blind, multicentre, placebo-controlled studies in patients 18 years of age or older with moderately to severely active psoriatic arthritis. All patients had active psoriatic arthritis for at least 6 months based upon the Classification Criteria for Psoriatic Arthritis (CASPAR), at least 3 tender joints and at least 3 swollen joints, and active plaque psoriasis or history of plaque psoriasis. For both studies, the primary endpoint was the proportion of patients who achieved an ACR20 response at week 12.
SELECT-PsA 1 was a 24-week trial in 1705 patients who had an inadequate response or intolerance to at least one non-biologic DMARD. At baseline, 1393 (82%) of patients were on at least one concomitant non-biologic DMARD; 1084 (64%) of patients received concomitant MTX only; and 311 (18%) of patients were on monotherapy. Patients received upadacitinib 15 mg or 30 mg once daily, adalimumab, or placebo. At week 24, all patients randomised to placebo were switched to upadacitinib 15 mg or 30 mg once daily in a blinded manner. SELECT-PsA 1 included a long-term extension for up to 5 years.
SELECT-PsA 2 was a 24-week trial in 642 patients who had an inadequate response or intolerance to at least one biologic DMARD. At baseline, 296 (46%) of patients were on at least one concomitant non-biologic DMARD; 222 (35%) of patients received concomitant MTX only; and 345 (54%) of patients were on monotherapy. Patients received upadacitinib 15 mg or 30 mg once daily or placebo. At week 24, all patients randomised to placebo were switched to upadacitinib 15 mg or 30 mg once daily in a blinded manner. SELECT-PsA 2 included a long-term extension for up to 3 years.
Clinical response: In both studies, a statistically significant greater proportion of patients treated with upadacitinib 15 mg achieved ACR20 response compared to placebo at week 12 (Table 5). Time to onset of efficacy was rapid across measures with greater responses seen as early as week 2 for ACR20.
Treatment with upadacitinib 15 mg resulted in improvements in individual ACR components, including tender/painful and swollen joint counts, patient and physician global assessments, HAQ-DI, pain assessment, and hsCRP compared to placebo.
In SELECT-PsA 1, upadacitinib 15 mg achieved non-inferiority compared to adalimumab in the proportion of patients achieving ACR20 response at week 12; however, superiority to adalimumab could not be demonstrated.
In both studies, consistent responses were observed alone or in combination with methotrexate for primary and key secondary endpoints.
The efficacy of upadacitinib 15 mg was demonstrated regardless of subgroups evaluated including baseline BMI, baseline hsCRP, and number of prior non-biologic DMARDs (≤1 or >1). (See Table 5.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageRadiographic response: In SELECT-PsA 1, inhibition of progression of structural damage was assessed radiographically and expressed as the change from baseline in modified Total Sharp Score (mTSS) and its components, the erosion score and the joint space narrowing score, at week 24.
Treatment with upadacitinib 15 mg resulted in statistically significant greater inhibition of the progression of structural joint damage compared to placebo at week 24 (Table 6). Erosion and joint space narrowing scores were consistent with the overall scores. The proportion of patients with no radiographic progression (mTSS change ≤0.5) was higher with upadacitinib 15 mg compared to placebo at week 24. (See Table 6.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePhysical function response and health-related outcomes: In SELECT-PsA 1, patients treated with upadacitinib 15 mg showed statistically significant improvement from baseline in physical function as assessed by HAQ-DI at week 12 (-0.42 [95% CI: -0.47, -0.37]) compared to placebo (-0.14 [95% CI: -0.18, -0.09]); improvement in patients treated with adalimumab was -0.34 (95% CI: -0.38, -0.29). In SELECT-PsA 2, patients treated with upadacitinib 15 mg showed statistically significant improvement from baseline in HAQ-DI at week 12 (-0.30 [95% CI: -0.37, -0.24]) compared to placebo (-0.10 [95% CI: -0.16, -0.03]). Improvement in physical function was maintained through week 56 in both studies.
Health-related quality of life was assessed by SF-36v2. In both studies, patients receiving upadacitinib 15 mg experienced statistically significant greater improvement from baseline in the Physical Component Summary score compared to placebo at week 12. Improvements from baseline were maintained through week 56 in both studies.
Patients receiving upadacitinib 15 mg experienced statistically significant improvement from baseline in fatigue, as measured by FACIT-F score, at week 12 compared to placebo in both studies. Improvements from baseline were maintained through week 56 in both studies.
At baseline, psoriatic spondylitis was reported in 31% and 34% of patients in SELECT-PsA 1 and SELECT-PsA 2, respectively. Patients with psoriatic spondylitis treated with upadacitinib 15 mg showed improvements from baseline in Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) scores compared to placebo at week 24. Improvements from baseline were maintained through week 56 in both studies.
Axial spondyloarthritis: Non-radiographic axial spondyloarthritis: The efficacy and safety of upadacitinib 15 mg once daily were assessed in a randomised, double-blind, multicentre, placebo-controlled study in patients 18 years of age or older with active non-radiographic axial spondyloarthritis. Study SELECT-AXIS 2 (nr-axSpA) was a 52-week placebo-controlled trial in 314 patients with active non-radiographic axial spondyloarthritis with an inadequate response to at least two NSAIDs or intolerance to or contraindication for NSAIDs. Patients must have had objective signs of inflammation indicated by elevated C-reactive protein (CRP) (defined as > upper limit of normal), and/or sacroiliitis on magnetic resonance imaging (MRI), and no definitive radiographic evidence of structural damage on sacroiliac joints. Patients had active disease as defined by the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥4, and a Patient's Assessment of Total Back Pain score ≥4 based on a 0-10 numerical rating scale (NRS) at the Screening and Baseline Visits. At baseline, patients had symptoms of non-radiographic axial spondyloarthritis for an average of 9.1 years and 29.1% of the patients were on a concomitant csDMARD. 32.9% of the patients had an inadequate response or intolerance to bDMARD therapy. Patients received upadacitinib 15 mg once daily or placebo. At week 52, all patients randomised to placebo were switched to upadacitinib 15 mg once daily. The primary endpoint was the proportion of patients achieving an Assessment of SpondyloArthritis international Society 40 (ASAS40) response at week 14. The study included a long term extension for up to 2 years. So far, only efficacy data up to week 14 are available and presented.
Clinical response: In SELECT-AXIS 2 (nr-axSpA), a significantly greater proportion of patients treated with upadacitinib 15 mg achieved an ASAS40 response compared to placebo at week 14 (Table 7). A numerical difference between treatment groups was observed at all timepoints from week 2 to week 14.
Treatment with upadacitinib 15 mg resulted in improvements in individual ASAS components (patient global assessment of disease activity, total back pain assessment, inflammation, and function) and other measures of disease activity, including hsCRP, compared to placebo at week 14.
The efficacy of upadacitinib 15 mg was demonstrated across subgroups including gender, baseline BMI, symptom duration of non-radiographic axial spondyloarthritis, baseline hsCRP, MRI sacroiliitis, and prior use of bDMARDs. (See Table 7.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePhysical function response and health-related outcomes: Patients treated with upadacitinib 15 mg showed significant improvement in physical function from baseline compared to placebo as assessed by the BASFI at week 14.
Patients treated with upadacitinib 15 mg showed significant improvements in total back pain and nocturnal back pain compared to placebo at week 14.
Patients treated with upadacitinib 15 mg showed significant improvements in health-related quality of life and overall health as measured by ASQoL and ASAS Health Index, respectively, compared to placebo at week 14.
Objective measure of inflammation: Signs of inflammation were assessed by MRI and expressed as change from baseline in the Spondyloarthritis Research Consortium of Canada (SPARCC) score of the sacroiliac joints. At week 14, significant improvement of inflammatory signs in the sacroiliac joint was observed in patients treated with upadacitinib 15 mg compared to placebo.
Ankylosing spondylitis (AS, radiographic axial spondyloarthritis): The efficacy and safety of upadacitinib 15 mg once daily were assessed in two randomised, double-blind, multicentre, placebo-controlled studies in patients 18 years of age or older with active ankylosing spondylitis based upon the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥4 and Patient's Assessment of Total Back Pain score ≥4. Both studies included a long-term extension for up to 2 years.
SELECT-AXIS 1 was a 14-week placebo-controlled trial in 187 ankylosing spondylitis patients with an inadequate response to at least two NSAIDs or intolerance to or contraindication for NSAIDs and had no previous exposure to biologic DMARDs. At baseline, patients had symptoms of ankylosing spondylitis for an average of 14.4 years and approximately 16% of the patients were on a concomitant csDMARD. Patients received upadacitinib 15 mg once daily or placebo. At week 14, all patients randomised to placebo were switched to upadacitinib 15 mg once daily. The primary endpoint was the proportion of patients achieving an Assessment of SpondyloArthritis international Society 40 (ASAS40) response at week 14.
SELECT-AXIS 2 (AS) was a 14-week placebo-controlled trial in 420 ankylosing spondylitis patients with prior exposure to bDMARDs (77.4% had lack of efficacy to either a TNF inhibitor or interleukin 17 inhibitor (IL-17i); 30.2% had intolerance; 12.9% had prior exposure but not lack of efficacy to two bDMARDs). At baseline, patients had symptoms of ankylosing spondylitis for an average of 12.8 years and approximately 31% of the patients were on a concomitant csDMARD. Patients received upadacitinib 15 mg once daily or placebo. At week 14, all patients randomised to placebo were switched to upadacitinib 15 mg once daily. The primary endpoint was the proportion of patients achieving an Assessment of SpondyloArthritis international Society 40 (ASAS40) response at week 14.
Clinical response: In both studies, a significantly greater proportion of patients treated with upadacitinib 15 mg achieved an ASAS40 response compared to placebo at week 14 (Table 8). A numerical difference between treatment groups was observed from week 2 in SELECT-AXIS 1 and week 4 in SELECT-AXIS 2 (AS) for ASAS40.
Treatment with upadacitinib 15 mg resulted in improvements in individual ASAS components (patient global assessment of disease activity, total back pain assessment, inflammation, and function) and other measures of disease activity, including hsCRP, at week 14 compared to placebo.
The efficacy of upadacitinib 15 mg was demonstrated regardless of subgroups evaluated including gender, baseline BMI, symptom duration of AS, baseline hsCRP, and prior use of bDMARDs. (See Table 8.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn SELECT-AXIS 1, efficacy was maintained through 2 years as assessed by the endpoints presented in Table 8 as previously mentioned.
Physical function response and health-related outcomes: In both studies, patients treated with upadacitinib 15 mg showed significant improvement in physical function from baseline compared to placebo as assessed by the Bath Ankylosing Spondylitis Functional Index (BASFI) change from baseline at week 14. In SELECT-AXIS 1, improvement in BASFI was maintained through 2 years.
In SELECT-AXIS 2 (AS), patients treated with upadacitinib 15 mg showed significant improvements in total back pain and nocturnal back pain compared to placebo at week 14.
In SELECT-AXIS 2 (AS), patients treated with upadacitinib 15 mg showed significant improvements in health-related quality of life and overall health as measured by ASQoL and ASAS Health Index, respectively, compared to placebo at week 14.
Enthesitis: In SELECT-AXIS 2 (AS), patients with pre-existing enthesitis (n=310) treated with upadacitinib 15 mg showed significant improvement in enthesitis compared to placebo as measured by change from baseline in Maastricht Ankylosing Spondylitis Enthesitis Score (MASES) at week 14.
Spinal mobility: In SELECT-AXIS 2 (AS), patients treated with upadacitinib 15 mg showed significant improvement in spinal mobility compared to placebo as measured by change from baseline in Bath Ankylosing Spondylitis Metrology Index (BASMI) at week 14.
Objective measure of inflammation: Signs of inflammation were assessed by MRI and expressed as change from baseline in the SPARCC score for spine. In both studies at week 14, significant improvement of inflammatory signs in the spine was observed in patients treated with upadacitinib 15 mg compared to placebo. In SELECT-AXIS 1, improvement in inflammation as assessed by MRI was maintained through 2 years.
Atopic dermatitis: The efficacy and safety of upadacitinib 15 mg and 30 mg once daily was assessed in three Phase 3 randomised, double-blind, multicentre studies (MEASURE UP 1, MEASURE UP 2 and AD UP) in a total of 2584 patients (12 years of age and older). Upadacitinib was evaluated in 344 adolescent and 2240 adult patients with moderate to severe atopic dermatitis (AD) not adequately controlled by topical medication(s). At baseline, patients had to have all the following: an Investigator's Global Assessment (vIGA-AD) score ≥3 in the overall assessment of AD (erythema, induration/papulation, and oozing/crusting) on an increasing severity scale of 0 to 4, an Eczema Area and Severity Index (EASI) score ≥16 (composite score assessing extent and severity of erythema, oedema/papulation, scratches and lichenification across 4 different body sites), a minimum body surface area (BSA) involvement of ≥10%, and weekly average Worst Pruritus Numerical Rating Scale (NRS) ≥4.
In all three studies, patients received upadacitinib once daily doses of 15 mg, 30 mg, or matching placebo for 16 weeks. In the AD UP study, patients also received concomitant topical corticosteroids (TCS). Following completion of the double blinded period, patients originally randomised to upadacitinib were to continue receiving the same dose until week 260. Patients in the placebo group were re-randomised in a 1:1 ratio to receive upadacitinib 15 mg or 30 mg until week 260.
Baseline characteristics: In the monotherapy studies (MEASURE UP 1 and 2), 50.0% of patients had a baseline vIGA-AD score of 3 (moderate) and 50.0% of patients had a baseline vIGA-AD of 4 (severe). The mean baseline EASI score was 29.3 and the mean baseline weekly average Worst Pruritus NRS was 7.3. In the concomitant TCS study (AD UP), 47.1% of patients had a baseline vIGA-AD score of 3 (moderate) and 52.9% of patients had a baseline vIGA-AD of 4 (severe). The mean baseline EASI score was 29.7 and the mean baseline weekly average Worst Pruritus NRS was 7.2.
Clinical response: Monotherapy (MEASURE UP 1 AND MEASURE UP 2) and Concomitant TCS (AD UP) studies: A significantly greater proportion of patients treated with upadacitinib 15 mg or 30 mg achieved vIGA-AD 0 or 1, EASI 75, or a ≥4-point improvement on the Worst Pruritus NRS compared to placebo at week 16. Rapid improvements in skin clearance and itch were also achieved (see Table 9).
Figure 1 shows the proportion of patients achieving an EASI 75 response and mean percent change from baseline in Worst Pruritus NRS, respectively up to week 16 for MEASURE UP 1 and 2. (See Table 9 and Figure 1.)
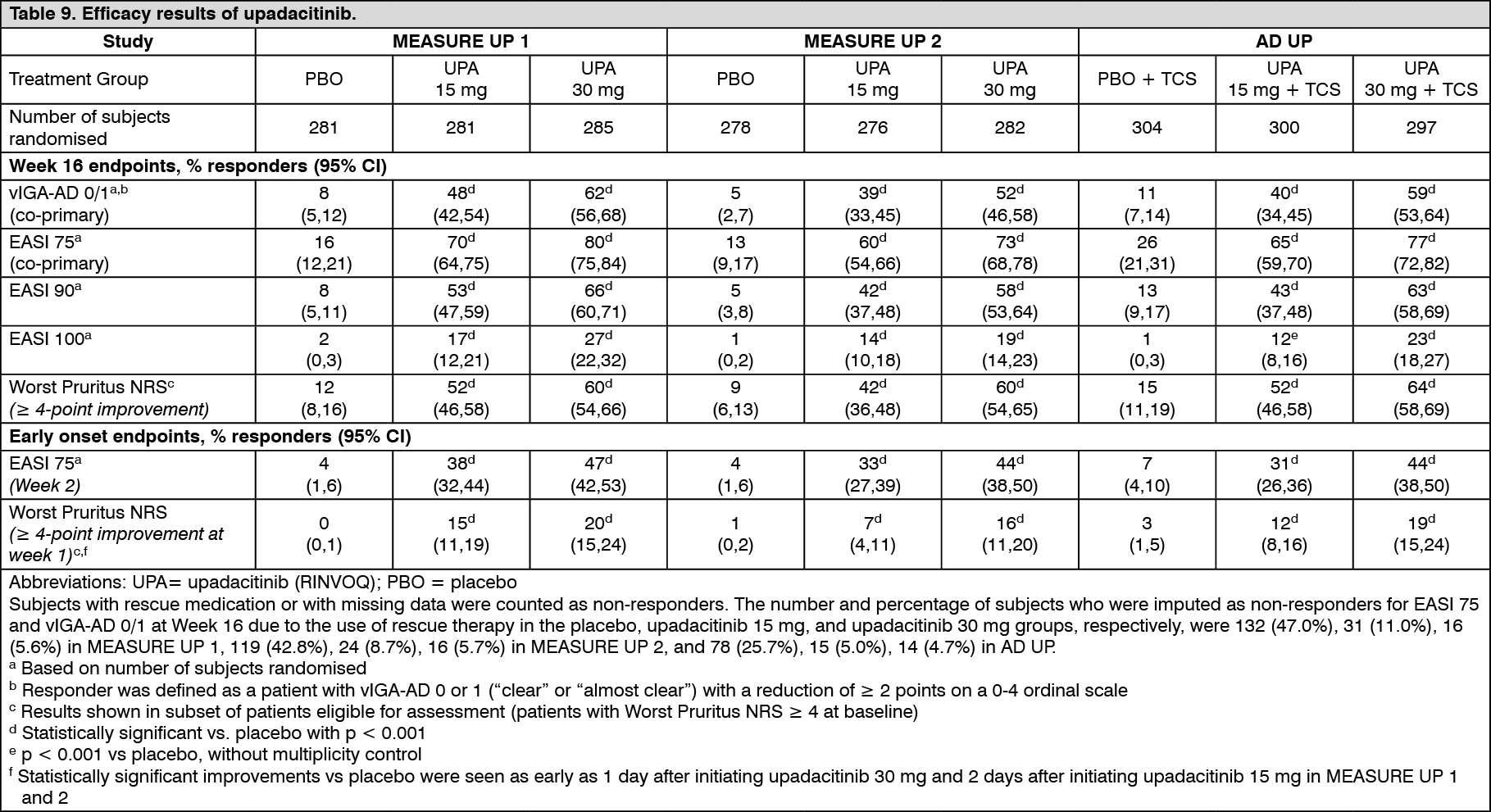 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageTreatment effects in subgroups (weight, age, gender, race, and prior systemic treatment with immunosuppressants) were consistent with the results in the overall study population.
Results at week 16 continued to be maintained through week 52 in patients treated with upadacitinib 15 mg or 30 mg.
Quality of life/patient-reported outcomes: (See Table 10.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageUlcerative colitis: The efficacy and safety of upadacitinib was evaluated in three multicentre, double-blind, placebo-controlled Phase 3 clinical studies: two replicate induction studies, UC-1 (U-ACHIEVE Induction) and UC-2 (U-ACCOMPLISH), and a maintenance study UC-3 (U-ACHIEVE Maintenance).
Disease activity was based on the adapted Mayo score (aMS, Mayo scoring system excluding Physician's Global Assessment), which ranged from 0 to 9 and has three subscores that were each scored 0 (normal) to 3 (most severe): stool frequency subscore (SFS), rectal bleeding subscore (RBS) and a centrally-reviewed endoscopy subscore (ES).
Induction studies (UC-1 and UC-2): In UC-1 and UC-2, 988 patients (473 and 515 patients, respectively) were randomised to upadacitinib 45 mg once daily or placebo for 8 weeks with a 2:1 treatment allocation ratio and included in the efficacy analysis. All enrolled patients had moderately to severely active ulcerative colitis defined as an aMS of 5 to 9 with an ES of 2 or 3 and demonstrated prior treatment failure including inadequate response, loss of response, or intolerance to prior conventional and/or biologic treatment. Prior treatment failure to at least 1 biologic therapy (prior biologic failure) was seen in 52% (246/473) and 51% (262/515) of patients, respectively. Previous treatment failure to conventional therapy but not biologics (without prior biologic failure) was seen in 48% (227/473) and 49% (253/515) of patients, respectively.
At baseline in UC-1 and UC-2, 39% and 37% of patients received corticosteroids, 1.1% and 0.6% of patients received methotrexate and 68% and 69% of patients received aminosalicylates. Concomitant use of thiopurine was not allowed during the studies. Patient disease activity was moderate (aMS ≥5, ≤7) in 61% and 60% of patients and severe (aMS >7) in 39% and 40% of patients.
The primary endpoint was clinical remission per aMS at week 8. Table 11 shows the primary and key secondary endpoints including clinical response, mucosal healing, histologic-endoscopic mucosal healing and deep mucosal healing. (See Table 11.)
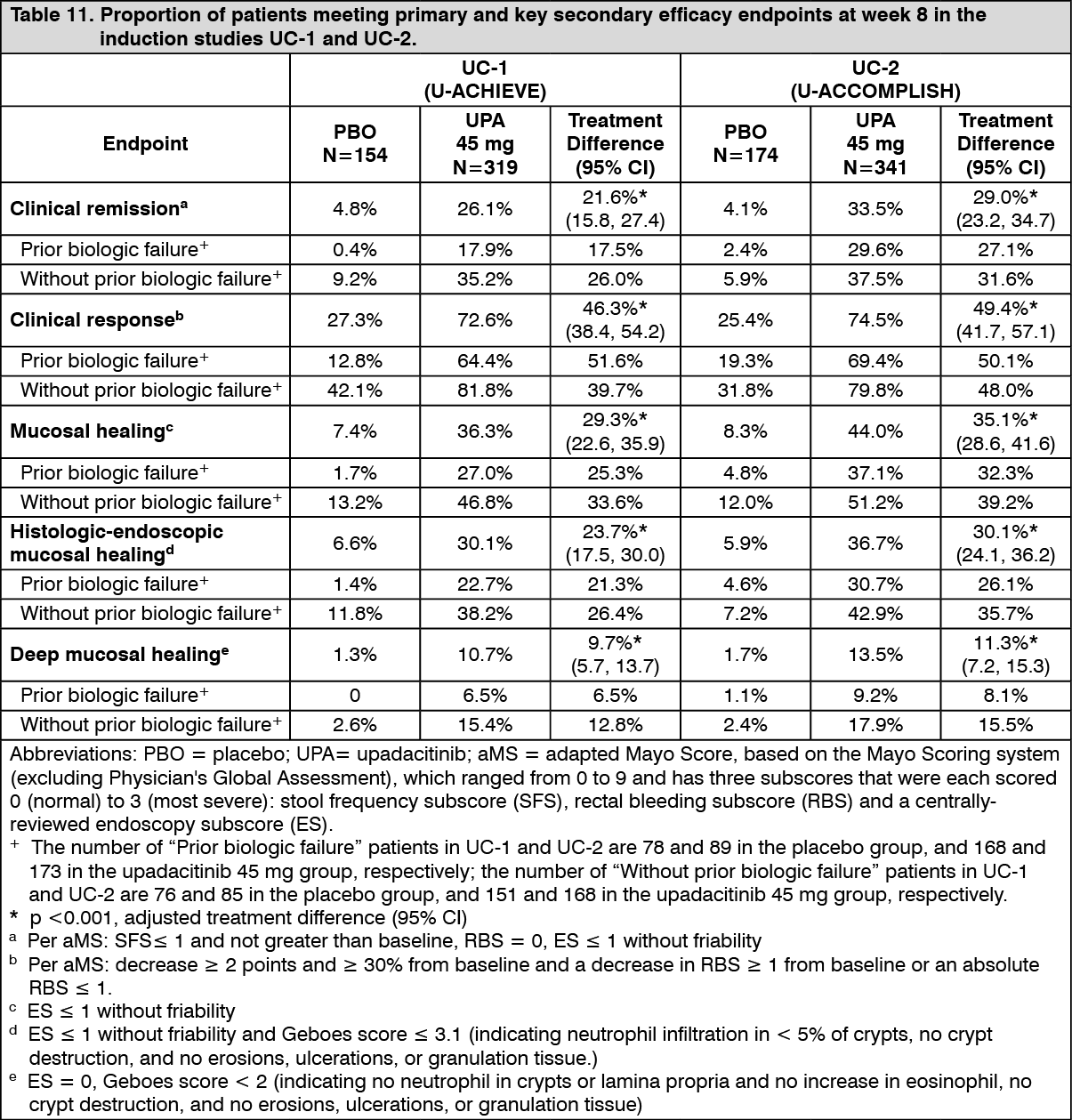 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageDisease activity and symptoms: The partial adapted Mayo score (paMS) is composed of SFS and RBS. Symptomatic response per paMS is defined as a decrease of ≥1 point and ≥30% from baseline and a decrease in RBS ≥1 or an absolute RBS ≤1. Statistically significant improvement compared to placebo per paMS was seen as early as week 2 (UC-1: 60.1% vs 27.3% and UC-2: 63.3% vs 25.9%).
Extended induction: A total of 125 patients in UC-1 and UC-2 who did not achieve clinical response after 8 weeks of treatment with upadacitinib 45 mg once daily entered an 8-week open-label extended induction period. After the treatment of an additional 8 weeks (16 weeks total) of upadacitinib 45 mg once daily, 48.3% of patients achieved clinical response per aMS. Among patients who responded to treatment of 16-week upadacitinib 45 mg once daily, 35.7% and 66.7% of patients maintained clinical response per aMS and 19.0% and 33.3% of patients achieved clinical remission per aMS at week 52 with maintenance treatment of upadacitinib 15 mg and 30 mg once daily, respectively.
Maintenance study (UC-3): The efficacy analysis for UC-3 was evaluated in 451 patients who achieved clinical response per aMS with 8-week upadacitinib 45 mg once daily induction treatment. Patients were randomised to receive upadacitinib 15 mg, 30 mg or placebo once daily for up to 52 weeks.
The primary endpoint was clinical remission per aMS at week 52. Table 12 shows the key secondary endpoints including maintenance of clinical remission, corticosteroid-free clinical remission, mucosal healing, histologic-endoscopic mucosal healing and deep mucosal healing. (See Table 12.)
 Click on icon to see table/diagram/image
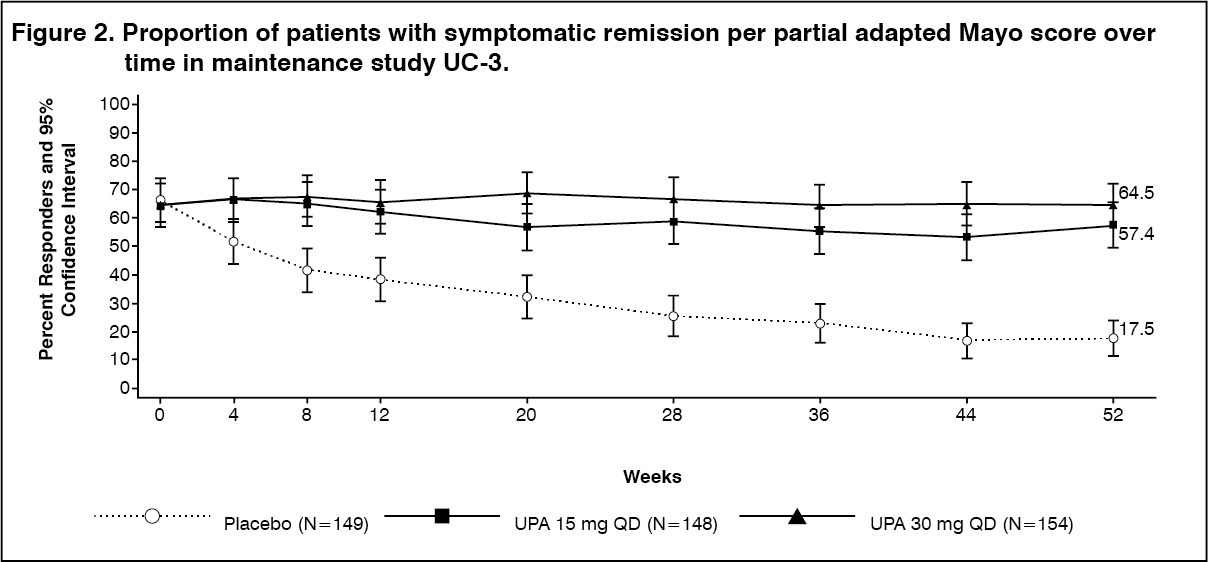
Click on icon to see table/diagram/imageDisease symptoms: Symptomatic remission per paMS, defined as SFS ≤1 and RBS = 0, was achieved over time through week 52 in more patients treated with both upadacitinib 15 mg and 30 mg once daily compared with placebo (see Figure 2).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageEndoscopic assessment: Endoscopic remission (normalisation of the endoscopic appearance of the mucosa) was defined as ES of 0. At week 8, a significantly greater proportion of patients treated with upadacitinib 45 mg once daily compared to placebo achieved endoscopic remission (UC-1: 13.7% vs 1.3%, UC-2: 18.2% vs 1.7%). In UC-3, a significantly greater proportion of patients treated with upadacitinib 15 mg and 30 mg once daily compared to placebo achieved endoscopic remission at week 52 (24.2% and 25.9% vs 5.6%). Maintenance of mucosal healing at week 52 (ES ≤1 without friability) was seen in a significantly greater proportion of patients treated with upadacitinib 15 mg and 30 mg once daily compared to placebo (61.6% and 69.5% vs 19.2%) among patients who achieved mucosal healing at the end of induction.
Quality of life: Patients treated with upadacitinib demonstrated significantly greater and clinically meaningful improvement in health-related quality of life measured by the Inflammatory Bowel Disease Questionnaire (IBDQ) total score compared to placebo. Improvements were seen in all 4 domain scores: systemic symptoms (including fatigue), social function, emotional function and bowel symptoms (including abdominal pain and bowel urgency). Changes in IBDQ total score at week 8 from baseline with upadacitinib 45 mg once daily compared to placebo were 55.3 and 21.7 in UC-1 and 52.2 and 21.1 in UC-2, respectively. Changes in IBDQ total score at week 52 from baseline were 49.2, 58.9 and 17.9 in patients treated with upadacitinib 15 mg, 30 mg once daily and placebo, respectively.
Crohn's disease: The efficacy and safety of upadacitinib was evaluated in three multicenter, double-blind, placebo-controlled Phase 3 studies: two induction studies, CD-1 (U-EXCEED) and CD-2 (U-EXCEL), followed by a 52-week maintenance treatment and long-term extension study, CD-3 (U-ENDURE).
The co-primary endpoints were clinical remission and endoscopic response at week 12 for CD-1 and CD-2, and at week 52 for CD-3.
Enrolled patients were 18 to 75 years of age with moderately to severely active Crohn's disease (CD), defined as an average daily very soft or liquid stool frequency (SF) ≥4 and/or average daily abdominal pain score (APS) ≥2, and a centrally-reviewed Simple Endoscopic Score for CD (SES-CD) of ≥6, or ≥4 for isolated ileal disease, excluding the narrowing component. Patients with symptomatic bowel strictures were excluded from CD studies.
Induction studies (CD-1 and CD-2): In CD-1 and CD-2, 1021 patients (495 and 526 patients, respectively) were randomised to upadacitinib 45 mg once daily or placebo for 12 weeks with a 2:1 treatment allocation ratio.
In CD-1, all patients had inadequate response or were intolerant to treatment with one or more biologic therapies (prior biologic failure). Of these patients, 61% (301/495) had inadequate response or were intolerant to two or more biologic therapies.
In CD-2, 45% (239/526) patients had an inadequate response or were intolerant to treatment with one or more biologic therapies (prior biologic failure), and 55% (287/526) had an inadequate response or were intolerant to treatment with conventional therapies but not to biologic therapy (without prior biologic failure).
At baseline in CD-1 and CD-2, 34% and 36% of patients received corticosteroids, 7% and 3% of patients received immunomodulators, and 15% and 25% of patients received aminosalicylates.
In both studies, patients receiving corticosteroids at baseline initiated a corticosteroid taper regimen starting at week 4.
Both studies included a 12-week extended treatment period with upadacitinib 30 mg once daily for patients who received upadacitinib 45 mg once daily and did not achieve clinical response per SF/APS (≥30% decrease in average daily very soft or liquid SF and/or ≥30% decrease in average daily APS and neither greater than baseline) at week 12.
Clinical disease activity and symptoms: In CD-1 and CD-2, a significantly greater proportion of patients treated with upadacitinib 45 mg achieved the co-primary endpoint of clinical remission at week 12 compared to placebo (Table 13). Onset of efficacy was rapid and achieved as early as week 2 (Table 13).
In both studies, patients receiving upadacitinib 45 mg experienced significantly greater improvement from baseline in fatigue, as measured by FACIT-F score at week 12 compared to placebo.
Endoscopic assessment: In CD-1 and CD-2, a significantly greater proportion of patients treated with upadacitinib 45 mg achieved the co-primary endpoint of endoscopic response at week 12 compared to placebo (Table 13). In CD-1 and CD-2, a greater proportion of patients treated with upadacitinib 45 mg (14% and 19%, respectively) compared to placebo (0% and 5%, respectively) achieved SES-CD 0-2. (See Table 13.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageMaintenance study (CD-3): The efficacy analysis for CD-3 evaluated 502 patients who achieved clinical response per SF/APS with the 12-week upadacitinib 45 mg once daily induction treatment. Patients were re-randomised to receive a maintenance regimen of either upadacitinib 15 mg or 30 mg once daily or placebo for 52 weeks.
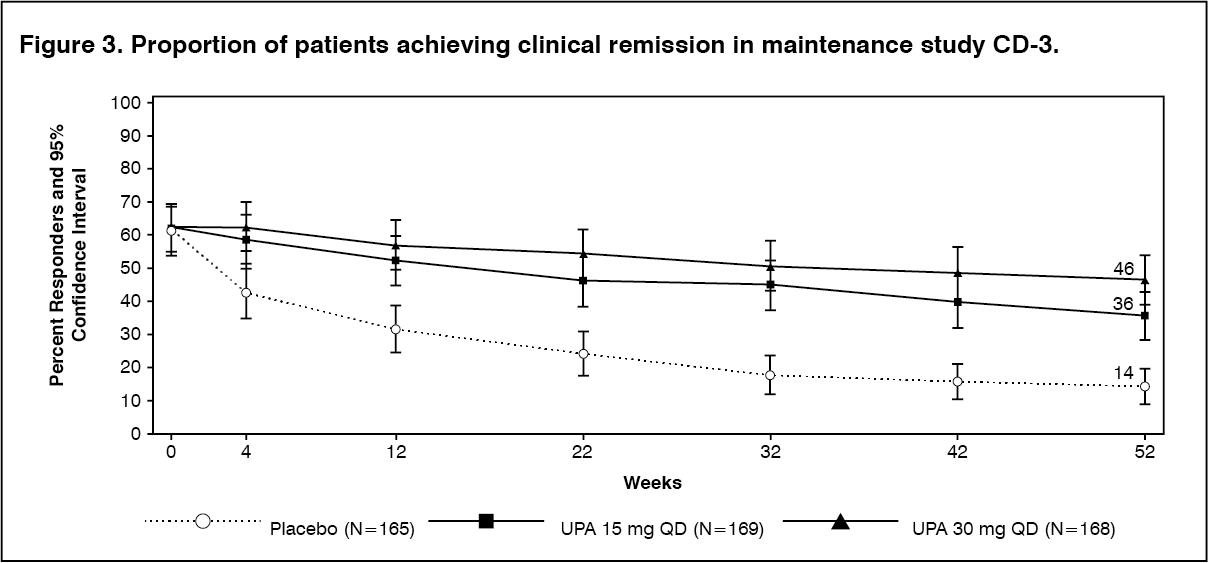
Clinical disease activity and symptoms: A significantly greater proportion of patients treated with upadacitinib 15 mg and 30 mg achieved the co-primary endpoint of clinical remission at week 52 compared to placebo (Figure 3, Table 14). (See Figure 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePatients receiving upadacitinib 30 mg experienced significantly greater improvement from baseline in fatigue, as measured by FACIT-F score at week 52 compared to placebo. (See Table 14.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePatients who were not in clinical response per SF/APS to upadacitinib induction at week 12 in CD-1 and CD-2 (122 patients) received upadacitinib 30 mg once daily for an additional 12 weeks. Of these patients, 53% achieved clinical response at week 24. Of the patients who responded to the extended treatment period and continued to receive maintenance treatment with upadacitinib 30 mg, 25% achieved clinical remission and 22% achieved endoscopic response at week 52.
Endoscopic assessment: In CD-3, a significantly greater proportion of patients treated with upadacitinib 15 mg and 30 mg achieved the co-primary endpoint of endoscopic response at week 52 compared to placebo (Table 14). In addition to the endoscopic endpoints described in Table 14, a greater proportion of patients treated with upadacitinib 15 mg and 30 mg (11% and 21%, respectively) compared to placebo (3%) achieved SES-CD 0-2 at week 52. Corticosteroid-free endoscopic remission among patients on steroid at baseline was achieved in a greater proportion of patients treated with upadacitinib 15 mg and 30 mg (17% and 25%, respectively) compared to placebo (3%) at week 52.
Resolution of extra-intestinal manifestations: Resolution of extra-intestinal manifestations was observed in a greater proportion of patients treated with upadacitinib 15 mg (25%) and a significantly greater proportion of patients treated with upadacitinib 30 mg (36%) compared to placebo (15%) at week 52.
Rescue treatment: In CD-3, patients who demonstrated inadequate response or lost response during maintenance were eligible to receive rescue treatment with upadacitinib 30 mg. Of the patients who were randomised to upadacitinib 15 mg group and received rescue treatment of upadacitinib 30 mg for at least 12 weeks, 84% (76/90) achieved clinical response per SF/APS and 48% (43/90) achieved clinical remission 12 weeks after initiating rescue.
Health-related quality of life outcomes: Patients treated with upadacitinib achieved greater improvement in health-related quality of life (HRQOL) measured by the Inflammatory Bowel Disease Questionnaire (IBDQ) total score compared to placebo. Improvements were seen in all 4 domain scores: systemic symptoms (including fatigue) and bowel symptoms (including abdominal pain and bowel urgency), as well as social and emotional functioning. Changes from baseline in IBDQ total score at week 12 with upadacitinib 45 mg once daily compared to placebo were 46.0 and 21.6 in CD-1 and 46.3 and 24.4 in CD-2, respectively. Changes in IBDQ total score at week 52 from baseline were 59.3, 64.5 and 46.4 in patients treated with upadacitinib 15 mg, 30 mg once daily and placebo, respectively.
Paediatric population: A total of 344 adolescents aged 12 to 17 years with moderate to severe atopic dermatitis were randomised across the three Phase 3 studies to receive either 15 mg (N=114) or 30 mg (N=114) upadacitinib or matching placebo (N=116), in monotherapy or combination with topical corticosteroids. Efficacy was consistent between the adolescents and adults. The safety profile in adolescents was generally similar to that in adults, with dose-dependent increases in the rate of some adverse events, including neutropaenia and herpes zoster. At both doses, the rate of neutropaenia was slightly increased in adolescents compared to adults. The rate of herpes zoster in adolescents at the 30 mg dose was comparable to that in adults. The safety and efficacy of the 30 mg dose in adolescents are still being investigated. (See Table 15.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePharmacokinetics: Upadacitinib plasma exposures are proportional to dose over the therapeutic dose range. Steady-state plasma concentrations are achieved within 4 days with minimal accumulation after multiple once daily administrations.
Absorption: Following oral administration of upadacitinib prolonged-release formulation, upadacitinib is absorbed with a median Tmax of 2 to 4 hours. Coadministration of upadacitinib with a high-fat meal had no clinically relevant effect on upadacitinib exposures (increased AUC by 29% and Cmax by 39% to 60%). In clinical trials, upadacitinib was administered without regard to meals (see Dosage & Administration). In vitro, upadacitinib is a substrate for the efflux transporters P-gp and BCRP.
Distribution: Upadacitinib is 52% bound to plasma proteins. Upadacitinib partitions similarly between plasma and blood cellular components, as indicated by the blood to plasma ratio of 1.0.
Metabolism: Upadacitinib metabolism is mediated by CYP3A4 with a potential minor contribution from CYP2D6. The pharmacologic activity of upadacitinib is attributed to the parent molecule. In a human radiolabeled study, unchanged upadacitinib accounted for 79% of the total radioactivity in plasma while the main metabolite (product of monooxidation followed by glucuronidation) accounted for 13% of the total plasma radioactivity. No active metabolites have been identified for upadacitinib.
Elimination: Following single dose administration of [14C]-upadacitinib immediate-release solution, upadacitinib was eliminated predominantly as the unchanged parent substance in urine (24%) and faeces (38%). Approximately 34% of upadacitinib dose was excreted as metabolites. Upadacitinib mean terminal elimination half-life ranged from 9 to 14 hours.
Special populations: Renal impairment: Upadacitinib AUC was 18%, 33%, and 44% higher in subjects with mild (estimated glomerular filtration rate 60 to 89 ml/min/1.73 m2), moderate (estimated glomerular filtration rate 30 to 59 ml/min/1.73 m2), and severe (estimated glomerular filtration rate 15 to 29 ml/min/1.73 m2) renal impairment, respectively, compared to subjects with normal renal function. Upadacitinib Cmax was similar in subjects with normal and impaired renal function. Mild or moderate renal impairment has no clinically relevant effect on upadacitinib exposure (see Dosage & Administration).
Hepatic impairment: Mild (Child-Pugh A) and moderate (Child-Pugh B) hepatic impairment has no clinically relevant effect on upadacitinib exposure. Upadacitinib AUC was 28% and 24% higher in subjects with mild and moderate hepatic impairment, respectively, compared to subjects with normal liver function. Upadacitinib Cmax was unchanged in subjects with mild hepatic impairment and 43% higher in subjects with moderate hepatic impairment compared to subjects with normal liver function. Upadacitinib was not studied in patients with severe (Child-Pugh C) hepatic impairment.
Paediatric population: The pharmacokinetics of upadacitinib have not yet been evaluated in paediatric patients with rheumatoid arthritis, psoriatic arthritis, axial spondyloarthritis, ulcerative colitis, and Crohn's disease (see Dosage & Administration).
Upadacitinib pharmacokinetics and steady-state concentrations are similar for adults and adolescents 12 to 17 years of age with atopic dermatitis. The posology in adolescent patients 30 kg to <40 kg was determined using population pharmacokinetic modelling and simulation.
The pharmacokinetics of upadacitinib in paediatric patients (<12 years of age) with atopic dermatitis have not been established.
Intrinsic factors: Age, sex, body weight, race, and ethnicity did not have a clinically meaningful effect on upadacitinib exposure. Upadacitinib pharmacokinetics are consistent between rheumatoid arthritis, psoriatic arthritis, axial spondyloarthritis, atopic dermatitis, ulcerative colitis, and Crohn's disease patients.
Toxicology: Preclinical safety data: Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology.
Upadacitinib, at exposures (based on AUC) approximately 4 and 10 times the clinical dose of 15 mg, 2 and 5 times the clinical dose of 30 mg, and 1.7 and 4 times the clinical dose of 45 mg in male and female Sprague-Dawley rats, respectively, was not carcinogenic in a 2-year carcinogenicity study in Sprague-Dawley rats. Upadacitinib was not carcinogenic in a 26-week carcinogenicity study in CByB6F1-Tg (HRAS)2Jic transgenic mice.
Upadacitinib was not mutagenic or genotoxic based on the results of in vitro and in vivo tests for gene mutations and chromosomal aberrations.
Upadacitinib had no effect on fertility in male or female rats at exposures up to approximately 17 and 34 times the maximum recommended human dose (MRHD) of 45 mg in males and females, respectively, on an AUC basis in a fertility and early embryonic development study. Dose-related increases in foetal resorptions associated with post-implantation losses in this fertility study in rats were attributed to the developmental/teratogenic effects of upadacitinib. No adverse effects were observed at exposures below clinical exposure (based on AUC). Post-implantation losses were observed at exposures 9 times the clinical exposure at the MRHD of 45 mg (based on AUC).
In animal embryo-foetal development studies, upadacitinib was teratogenic in both rats and rabbits. Upadacitinib resulted in increases in skeletal malformations in rats at 1.6, 0.8, and 0.6 times the clinical exposure (AUC-based) at the 15, 30, and 45 mg (MRHD) doses, respectively. In rabbits an increased incidence of cardiovascular malformations was observed at 15, 7.6, and 6 times the clinical exposure at the 15, 30, and 45 mg doses (AUC-based), respectively.
Following administration of upadacitinib to lactating rats, the concentrations of upadacitinib in milk over time generally paralleled those in plasma, with approximately 30-fold higher exposure in milk relative to maternal plasma. Approximately 97% of upadacitinib-related material in milk was the parent molecule, upadacitinib.