Pharmacotherapeutic group: Drugs for obstructive airway diseases, other systemic drugs for obstructive airway diseases.
ATC code: R03DX11.
Pharmacology: Pharmacodynamics: Mechanism of action: Tezepelumab is a monoclonal antibody (IgG2λ) directed against thymic stromal lymphopoietin (TSLP), preventing its interaction with the heterodimeric TSLP receptor. In asthma, both allergic and non-allergic triggers induce TSLP production. Blocking TSLP with tezepelumab reduces a broad spectrum of biomarkers and cytokines associated with airway inflammation (e.g. blood eosinophils, airway submucosal eosinophils, IgE, FeNO, IL-5, and IL-13); however, the mechanism of action of tezepelumab in asthma has not been definitively established.
Pharmacodynamic effects: Effect on blood eosinophils and inflammatory biomarkers and cytokines: In clinical trials, administration of tezepelumab 210 mg subcutaneously every 4 weeks reduced blood eosinophils counts, FeNO, IL-5 concentration, IL-13 concentration and serum IgE concentration from baseline compared with placebo. These markers were near maximal suppression after 2 weeks of treatment, except for IgE which declined more slowly. These effects were sustained throughout treatment.
Effect on eosinophils in the airway submucosa: In a clinical trial, administration of tezepelumab 210 mg subcutaneously every 4 weeks reduced submucosal eosinophil counts by 89% compared with a 25% reduction with placebo. Reduction was consistent regardless of baseline inflammatory biomarkers.
Immunogenicity: In NAVIGATOR, anti-drug antibodies (ADA) were detected at any time in 26 (4.9%) out of 527 patients who received tezepelumab at the recommended dosing regimen during the 52-week study period. Of these 26 patients, 10 patients (1.9% of patients treated with tezepelumab) developed treatment-emergent ADA and 1 patient (0.2% of patients treated with tezepelumab) developed neutralising antibodies. ADA titres were generally low and often transient. No evidence of ADA impact on pharmacokinetics, pharmacodynamics, efficacy, or safety was observed.
Clinical efficacy: The efficacy of tezepelumab was evaluated in two randomised, double-blind, parallel group, placebo-controlled clinical trials (PATHWAY and NAVIGATOR) of 52 weeks in duration involving a total of 1609 patients aged 12 years and older with severe asthma. In both trials, patients were enrolled without requiring a minimum baseline level of blood eosinophils or other inflammatory biomarkers (e.g. FeNO or IgE).
PATHWAY was a 52-week exacerbation trial which enrolled 550 patients (18 years of age and older) with severe, uncontrolled asthma to receive treatment with tezepelumab 70 mg subcutaneous Q4W, tezepelumab 210 mg subcutaneous Q4W, tezepelumab 280 mg subcutaneous Q2W or placebo. Patients were required to have a history of 2 or more asthma exacerbations requiring oral or systemic corticosteroid treatment or 1 asthma exacerbation resulting in hospitalisation in the past 12 months.
NAVIGATOR was a 52-week exacerbation trial which enrolled a total of 1061 patients (adults and adolescents 12 years of age and older) with severe, uncontrolled asthma to receive treatment with tezepelumab 210 mg subcutaneous Q4W or placebo. Patients were required to have a history of 2 or more asthma exacerbations requiring oral or systemic corticosteroid treatment or resulting in hospitalisation in the past 12 months.
In both PATHWAY and NAVIGATOR, patients were required to have an Asthma Control Questionnaire 6 (ACQ-6) score of 1.5 or more at screening, and reduced lung function at baseline (pre-bronchodilator FEV
1 below 80% predicted in adults, and below 90% predicted in adolescents). Patients were required to have been on regular treatment with medium- or high-dose inhaled corticosteroids (ICS) and at least one additional asthma control therapy with or without oral corticosteroids (OCS). High ICS dose was defined as > 500 mcg fluticasone propionate or equivalent per day. Medium ICS dose was defined as > 250 to 500 mcg fluticasone propionate or equivalent per day in PATHWAY and as 500 mcg fluticasone propionate or equivalent per day in NAVIGATOR. Patients continued background asthma therapy throughout the duration of the trials.
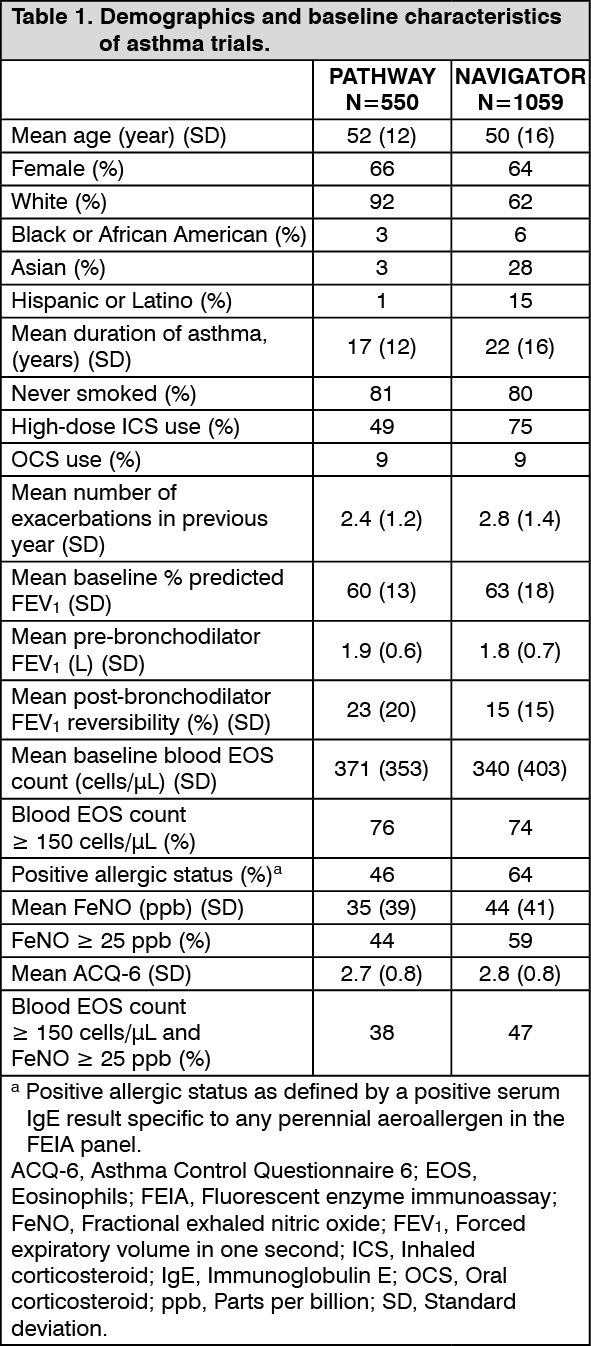
The demographics and baseline characteristics of these two trials are provided in Table 1 as follows. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The results summarised as follows are for the recommended tezepelumab 210 mg subcutaneous Q4W dosing regimen.
Exacerbations: The primary endpoint for PATHWAY and NAVIGATOR was the rate of severe asthma exacerbations measured over 52 weeks. Severe asthma exacerbations were defined as worsening of asthma requiring the use of or increase in oral or systemic corticosteroids for at least 3 days or a single depo-injection of corticosteroids, and/or emergency department visits requiring use of oral or systemic corticosteroids and/or hospitalisation.
In both PATHWAY and NAVIGATOR, patients receiving tezepelumab had significant reductions in the annualised rate of severe asthma exacerbations compared with placebo (Table 2 and Table 3). There were also fewer exacerbations requiring emergency room visits and/or hospitalisation in patients treated with tezepelumab compared with placebo. In PATHWAY and NAVIGATOR, severe asthma exacerbations requiring emergency room visits and/or hospitalisation were reduced by 85% and 79% with tezepelumab 210 mg subcutaneous Q4W, respectively. (See Tables 2 and 3.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Subgroup analysis: In NAVIGATOR, tezepelumab demonstrated a reduction in the rate of severe asthma exacerbations regardless of the baseline levels of blood eosinophils, FeNO, as well as allergic status (determined by a perennial aeroallergen specific IgE). Similar results were seen in PATHWAY. (See figure.)
In NAVIGATOR, reductions in the rate of severe asthma exacerbations were greater with increasing baseline blood eosinophil counts and FeNO values (rate ratio = 0.79 [95% CI: 0.48, 1.28] for patients with both baseline blood eosinophil count < 150 cells/μL and baseline FeNO < 25 ppb; rate ratio = 0.30 [95% CI: 0.23, 0.40] for patients with both baseline blood eosinophil count ≥ 150 cells/μL and baseline FeNO ≥ 25 ppb).
Click on icon to see table/diagram/image
Lung function: Change from baseline in FEV
1 was assessed as a secondary endpoint in NAVIGATOR. Compared with placebo, tezepelumab provided clinically meaningful improvements in the mean change from baseline in FEV
1 (Table 4).
Patient reported outcomes: Changes from baseline in ACQ-6, Standardised Asthma Quality of Life Questionnaire for ages 12 and older [AQLQ(S)+12] and weekly mean Asthma Symptom Diary (ASD) scores were assessed as secondary endpoints in NAVIGATOR. Severity of wheezing, shortness of breath, cough, and chest tightness were assessed twice daily (morning and evening). Night-time awakening and activity were assessed on a daily basis. The total ASD score was calculated as the mean of 10 items (Table 4).
Improvements in ACQ-6 and AQLQ(S)+12 were seen as early as 2 weeks and 4 weeks after administration of tezepelumab, respectively, and sustained through week 52 in both trials. (See Table 4.)
Click on icon to see table/diagram/image
Elderly patients (≥ 65 years of age): Of the 665 patients with asthma exposed to tezepelumab 210 mg subcutaneous Q4W in PATHWAY and NAVIGATOR, a total of 119 patients were 65 years of age or older, of which 32 patients were 75 years of age or older. Safety in these age groups were similar to the overall study population. Efficacy in these age groups were similar to the overall study population in NAVIGATOR. PATHWAY did not include sufficient numbers of patients aged 65 and over to determine efficacy in this age group.
Paediatric population: A total of 82 adolescents aged 12 to 17 with severe, uncontrolled asthma were enrolled in NAVIGATOR and received treatment with tezepelumab (n=41) or placebo (n=41). Of the 41 adolescents receiving treatment with tezepelumab, 15 were taking high-dose ICS at baseline. The annualised asthma exacerbation rate observed in adolescents treated with tezepelumab was 0.68 versus 0.97 for placebo (rate ratio 0.70; 95% CI 0.34, 1.46). The LS mean change from baseline for FEV
1 observed in adolescents treated with tezepelumab was 0.44 L versus 0.27 L for placebo (LS mean difference 0.17 L; 95% CI -0.01, 0.35). The pharmacodynamic responses in adolescents were generally similar to the overall study population.
Pharmacokinetics: The pharmacokinetics of tezepelumab were dose-proportional following subcutaneous administration over a dose range of 2.1 mg to 420 mg.
Absorption: Following a single subcutaneous administration, the maximum serum concentration was reached in approximately 3 to 10 days. Based on population pharmacokinetic analysis, the estimated absolute bioavailability was approximately 77%. There was no clinically relevant difference in bioavailability when administered to different injection sites (abdomen, thigh, or upper arm).
Distribution: Based on population pharmacokinetic analysis, central and peripheral volume of distribution of tezepelumab were 3.9 L and 2.2 L, respectively, for a 70 kg individual.
Metabolism: Tezepelumab is a human monoclonal antibody (IgG2λ) that is degraded by proteolytic enzymes widely distributed in the body and not metabolised by hepatic enzymes.
Elimination: As a human monoclonal antibody, tezepelumab is eliminated by intracellular catabolism and there is no evidence of target-mediated clearance. From population pharmacokinetic analysis, the estimated clearance for tezepelumab was 0.17 L/d for a 70 kg individual. The elimination half-life was approximately 26 days.
Special populations: Age, gender, race: Based on population pharmacokinetic analysis, age, gender and race had no clinically meaningful effects on the pharmacokinetics of tezepelumab.
Body weight: Based on population pharmacokinetic analysis, higher body weight was associated with lower exposure. However, the effect of body weight on exposure had no meaningful impact on efficacy or safety and does not require dose adjustment.
Paediatric patients: Based on the population pharmacokinetic analysis, there was no clinically meaningful age-related difference in the pharmacokinetics of tezepelumab between adults and adolescents aged 12 to 17 years. Tezepelumab has not been studied in children under 12 years of age (see Dosage & Administration).
Elderly patients (≥ 65 years of age): Based on population pharmacokinetic analysis, there was no clinically meaningful difference in the pharmacokinetics of tezepelumab between patients 65 years of age or older and younger patients.
Renal impairment: No formal clinical studies have been conducted to investigate the effect of renal impairment on tezepelumab. Based on population pharmacokinetic analysis, tezepelumab clearance was similar in patients with mild renal impairment (creatinine clearance 60 to < 90 mL/min), moderate renal impairment (creatinine clearance 30 to < 60 mL/min) and those with normal renal function (creatinine clearance ≥ 90 mL/min). Tezepelumab has not been studied in patients with severe renal impairment (creatinine clearance < 30 mL/min); however, tezepelumab is not cleared renally.
Hepatic impairment: No formal clinical studies have been conducted to investigate the effect of hepatic impairment on tezepelumab. IgG monoclonal antibodies are not primarily cleared via hepatic pathway; change in hepatic function is not expected to influence tezepelumab clearance. Based on population pharmacokinetic analysis, baseline hepatic function biomarkers (ALT, AST, and bilirubin) had no effect on tezepelumab clearance.
Toxicology: Preclinical safety data: Non-clinical data revealed no special hazard for humans based on repeated dose toxicity studies including safety pharmacology and fertility evaluations, and an ePPND (enhanced Pre- and Post-Natal Development) reproductive toxicity study in cynomolgus monkeys at doses of up to 300 mg/kg/week (producing exposures of greater than 100-times the clinical exposure at maximum recommended human dose [MRHD]).
Tezepelumab is excreted in milk in monkeys, although at low concentrations (< 1%).
Tezepelumab is a monoclonal antibody, as such genotoxicity and carcinogenicity studies have not been conducted.




 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image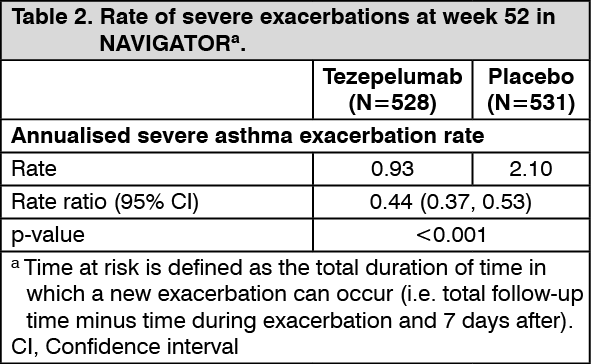 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image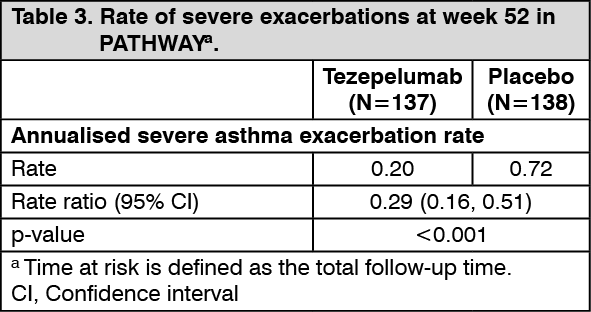 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image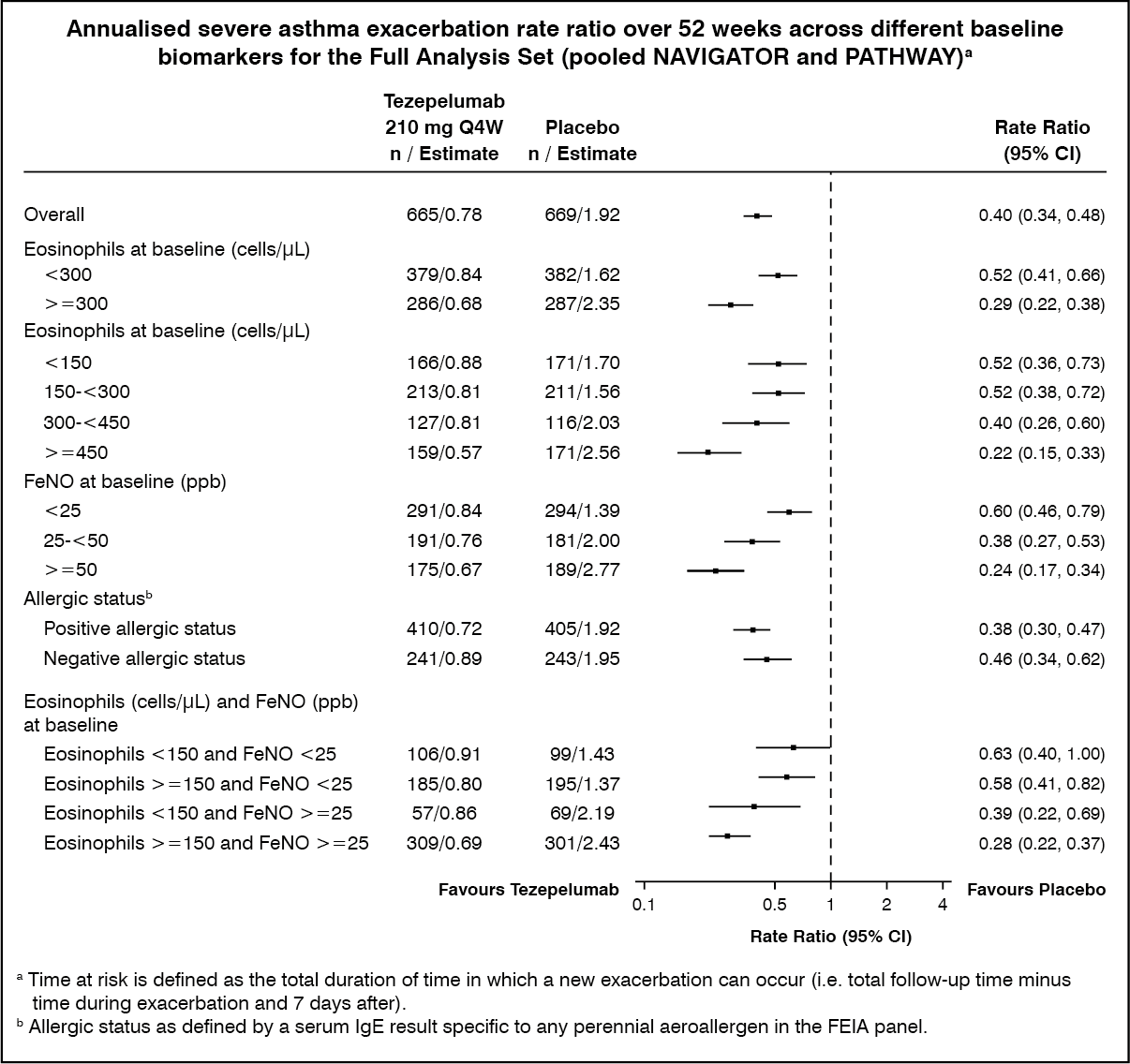 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image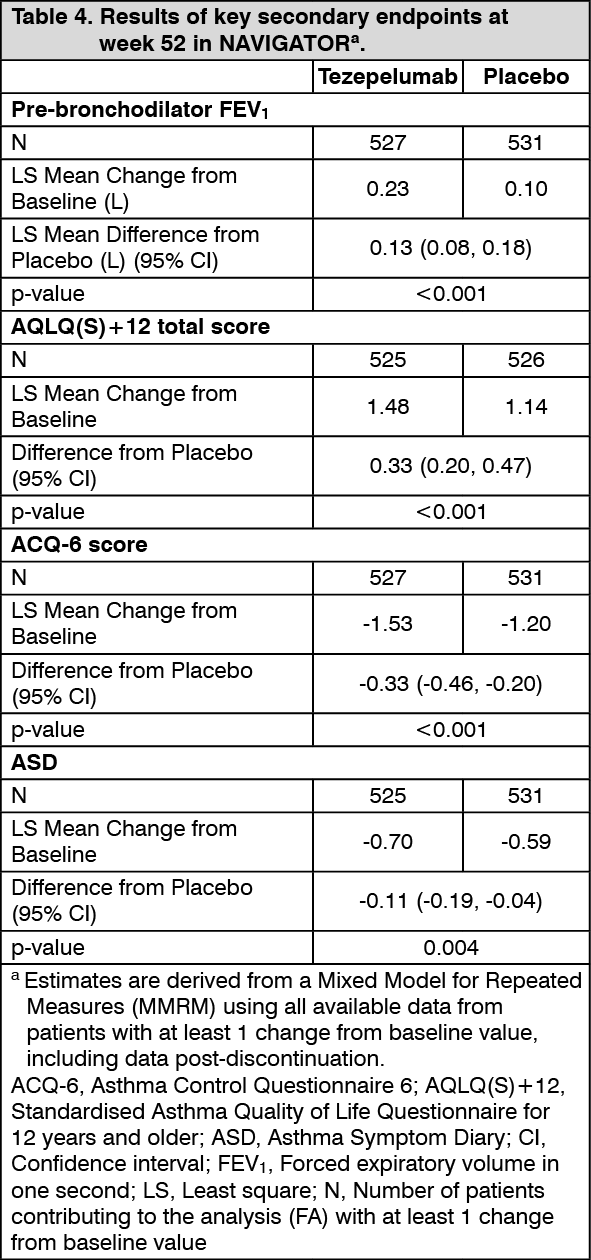 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out