Each capsule for oral use contains 0.5 mg dutasteride.
Excipients/Inactive Ingredients: Capsule contents: monodiglycerides of caprylic/capric acid; butylated hydroxytoluene.
Capsule shell: gelatin; glycerol; titanium dioxide (E171, CI 77891), iron oxide yellow (E172, CI 77492).
Medium chain triglycerides and lecithin as capsule lubricants.
Pharmacology: Pharmacodynamics: Dutasteride is a dual inhibitor of 5-alpha-reductase. It inhibits both type 1 and type 2, 5-alpha-reductase isoenzymes, which are responsible for the conversion of testosterone to 5-alpha-dihydrotestosterone (DHT). DHT is the androgen primarily responsible for hyperplasia of glandular prostatic tissue.
Effects on DHT/testosterone: The maximum effect of daily doses of Avodart on the reduction of DHT is dose-dependent and is observed within 1-2 weeks. After 1 week and 2 weeks of daily dosing of Avodart 0.5 mg, median serum DHT concentrations were reduced by 85% and 90%, respectively.
In BPH patients treated with 0.5 mg of dutasteride daily, the median decrease in DHT was 94% at 1 year and 93% at 2 years and the median increase in serum testosterone was 19% at both 1 and 2 years. This is an expected consequence of 5 alpha-reductase inhibition and did not result in any known adverse events.
Clinical Studies: Avodart Monotherapy: Dutasteride 0.5 mg/day or placebo was evaluated in 4,325 male subjects with enlarged prostates (greater than 30 cc) in three primary efficacy 2-year multicenter, placebo-controlled, double-blind studies.
In men with BPH, Avodart treats and prevents disease progression by reducing the risk of both acute urinary retention (AUR) and the need for surgical intervention (SI) and by providing statistically significant improvement of lower urinary tract symptoms (LUTS), maximum urinary flow rate (Qmax) and prostate volume relative to placebo. These improvements in LUTS, Qmax and prostate volume were seen through to 24 months, and LUTS and Qmax continued to improve for a further 2 years in open-label extension studies. In addition, reductions in prostate volume were sustained for a further 2 years in open-label extension studies.
Avodart and Tamsulosin Combination Therapy: Avodart 0.5 mg/day, tamsulosin 0.4 mg/day or the combination of Avodart 0.5 mg plus tamsulosin 0.4 mg was evaluated in 4,844 male subjects with enlarged prostates (greater than or equal to 30 cc) in a multicenter, double-blind, parallel group study over 2 years. The primary efficacy endpoint at 2 years of treatment was the level of improvement from baseline in the international prostate symptom score (IPSS).
After 2 years of treatment, combination therapy showed a statistically significant adjusted mean improvement in symptom scores from baseline of -6.2 units. The adjusted mean improvements in symptom scores observed with the individual therapies were -4.9 units for Avodart and -4.3 units for tamsulosin. The adjusted mean improvement in flow rate from baseline was 2.4 mL/sec for the combination, 1.9 mL/sec for Avodart and 0.9 mL/sec for tamsulosin. The adjusted mean improvement in BPH Impact Index (BII) from baseline was -2.1 units for the combination, -1.7 for Avodart and -1.5 for tamsulosin.
The reduction in total prostate volume and transition zone volume after 2 years of treatment was statistically significant for combination therapy compared to tamsulosin monotherapy alone.
The primary efficacy endpoint at 4 years of treatment was time to first event of AUR or BPH-related surgery. After 4 years of treatment, combination therapy statistically significantly reduced the risk of AUR or BPH-related surgery (65.8% reduction in risk p<0.001 [95% CI 54.7% to 74.1%]) compared to tamsulosin monotherapy. The incidence of AUR or BPH-related surgery by Year 4 was 4.2% for combination therapy and 11.9% for tamsulosin (p<0.001). Compared to Avodart monotherapy, combination therapy reduced the risk of AUR or BPH-related surgery by 19.6%; the difference between treatment groups was not significant (p=0.18 [95% CI ‑10.9% to 41.7%]). The incidence of AUR or BPH-related surgery by Year 4 was 4.2% for combination therapy and 5.2% for Avodart.
Clinical progression was defined as a composite of worsening symptoms, (IPSS), and BPH‑related events of AUR, incontinence, UTI, and renal insufficiency. Combination therapy was associated with a statistically significantly lower rate of clinical progression compared with tamsulosin (p<0.001, 44.1% risk reduction [95 % CI: 33.6% to 53.0%]) after 4 years. The rates of clinical progression for combination therapy, tamsulosin, and Avodart were: 12.6%, 21.5%, and 17.8%, respectively.
The statistically significant adjusted mean improvement in symptom scores (IPSS) from baseline was maintained from Year 2 to Year 4. At 4 years, the adjusted mean improvements in symptom scores observed were -6.3 units for combination therapy, -5.3 units for Avodart monotherapy and -3.8 units for tamsulosin monotherapy.
After 4 years of treatment, the adjusted mean improvement in flow rate (Qmax) from baseline was 2.4 mL/sec for combination therapy, 2.0 mL/sec for Avodart monotherapy and 0.7 mL/sec for tamsulosin monotherapy. Compared with tamsulosin, the adjusted mean improvement from baseline in Qmax was statistically significantly greater with combination therapy at each 6‑month assessment from Month 6 to Month 48 (p<0.001). Compared with Avodart, the adjusted mean improvement from baseline in Qmax was not statistically significantly different than with combination therapy (p=0.050 at Month 48).
Combination therapy was significantly superior (p<0.001) to tamsulosin monotherapy and to Avodart monotherapy for the improvement in health outcome parameters BII and BPH-related Health Status (BHS) at 4 years. The adjusted mean improvement in BII from baseline was -2.2 units for the combination, -1.8 for Avodart and -1.2 for tamsulosin. The adjusted mean improvement in BHS from baseline was -1.5 units for the combination, -1.3 for Avodart and -1.1 for tamsulosin.
The reduction in total prostate volume and transition zone volume after 4 years of treatment was statistically significant for combination therapy compared to tamsulosin monotherapy alone.
Cardiac failure: In a 4-year comparison of Avodart co-administered with tamsulosin and dutasteride or tamsulosin monotherapy in men with BPH (the CombAT study), the incidence of the composite term cardiac failure in the combination group (14/1,610, 0.9%) was higher than in either monotherapy group: Avodart, 4/1,623 (0.2%) and tamsulosin, 10/1,611, (0.6%). The relative risk estimate for time to first cardiac failure event was 3.57 [95% CI 1.17, 10.8] for combination treatment compared to Avodart monotherapy and 1.36 [95% CI 0.61, 3.07] compared to tamsulosin monotherapy.
In a 4-year chemoprevention comparison study of placebo and Avodart in 8,231 men aged 50 to 75, with a prior negative biopsy for prostate cancer and baseline PSA between 2.5 nanograms/mL and 10.0 nanograms/mL (the REDUCE study) there was a higher incidence of the composite term cardiac failure in subjects taking Avodart (30/4,105; 0.7%) versus placebo (16/4,126; 0.4%) for a relative risk estimate for time to first cardiac failure event of 1.91 [95% CI 1.04, 3.50]. In a post-hoc analysis of concomitant alpha blocker use, there was a higher incidence of the composite term cardiac failure in subjects taking Avodart and an alpha blocker concomitantly (12/1,152; 1.0%), compared to subjects not taking Avodart and an alpha blocker concomitantly: Avodart and no alpha blocker (18/2,953; 0.6%), placebo and an alpha blocker (1/1,399; <0.1%), placebo and no alpha blocker (15/2,727; 0.6%). No causal relationship between Avodart (alone or in combination with an alpha blocker) and cardiac failure has been established (see Precautions).
Prostate cancer and high grade tumours: In a 4-year comparison of placebo and Avodart in 8,231 men aged 50 to 75, with a prior negative biopsy for prostate cancer and baseline PSA between 2.5 nanograms/mL and 10.0 nanograms/mL (the REDUCE study), 6,706 subjects had prostate needle biopsy data available for analysis to determine Gleason Scores. There were 1,517 subjects diagnosed with prostate cancer in the study. The majority of biopsy-detectable prostate cancers in both treatment groups were diagnosed as low grade (Gleason 5-6). There was no difference in the incidence of Gleason 7-10 cancers (p=0.81).
There was a higher incidence of Gleason 8-10 prostate cancers in the Avodart group (n=29, 0.9%) compared to the placebo group (n=19, 0.6%) (p=0.15). In Years 1-2, the number of subjects with Gleason 8-10 cancers was similar in the Avodart group (n=17, 0.5%) and the placebo group (n=18, 0.5%). In Years 3-4, more Gleason 8-10 cancers were diagnosed in the Avodart group (n=12, 0.5%) compared with the placebo group (n=1, <0.1%) (p=0.0035). There are no data available on the effect of Avodart beyond 4 years in men at risk of prostate cancer. The percentage of subjects diagnosed with Gleason 8-10 cancers was consistent across study time periods (Years 1-2 and Years 3-4) in the Avodart group (0.5% in each time period), while in the placebo group, the percentage of subjects diagnosed with Gleason 8-10 cancers was lower during Years 3-4 than in Years 1-2 (<0.1% versus 0.5%, respectively). In a 4 year BPH study (CombAT) where there were no protocol-mandated biopsies and all diagnoses of prostate cancer were based on for-cause biopsies, the rates of Gleason 8-10 cancer were (n=8, 0.5%) for Avodart, (n=11, 0.7%) for tamsulosin and (n=5, 0.3%) for combination therapy (see Precautions).
The results of an epidemiological, population-based study (n=174,895) in community practice settings show that the use of 5-ARIs to treat BPH/LUTS is not associated with an increased risk of prostate cancer mortality (hazard ratio adjusted for competing risks: 0.85, 95% CI 0.72, 1.01) when compared with the use of alpha-blockers. Similar results were reported in an epidemiological study (n=13,892) of men with prostate cancer in the UK (adjusted hazard ratio for prostate cancer mortality for 5-ARI users versus non-users: 0.86; 95% CI 0.69, 1.06). A prospective cohort study, the Health Professional’s Follow-up study (n=38,058), also found that 5-ARI use was not associated with fatal prostate cancer (adjusted HR: 0.99; 95% CI 0.58, 1.69).
Effects on prostate specific antigen (PSA) and prostate cancer detection: In the REDUCE study, patients with a prior negative biopsy for prostate cancer and baseline PSA between 2.5 nanograms/mL and 10.0 nanograms/mL, Avodart treatment caused a decrease in mean serum PSA by approximately 50% after six months of treatment with a large variability (standard deviation of 30%) among patients. The PSA suppression observed at six months was similar in men who did or who did not develop biopsy-detectable prostate cancer during the study (see Precautions).
Incidence Of Breast Cancer: In BPH monotherapy clinical trials, providing 3,374 patient years of exposure to Avodart, there were 2 cases of male breast cancer reported in Avodart-treated patients, one after 10 weeks and one after 11 months of treatment and 1 case in a patient who received placebo. In subsequent clinical trials in BPH and 8,231 men aged 50 to 75, with a prior negative biopsy for prostate cancer and baseline PSA between 2.5 nanograms/mL and 10.0 nanograms/mL providing 17,489 patient years exposure to Avodart and 5,027 patient years exposure to Avodart and tamsulosin combination there were no reported breast cancer cases in any of the treatment groups.
Two case control, epidemiological studies, one conducted in a US (n=339 breast cancer cases and n=6,780 controls) and the other in a UK (n=338 breast cancer cases and n=3,930 controls) healthcare database, showed no increase in the risk of developing male breast cancer with the use of 5-ARIs (see Precautions). Results from the first study did not identify a positive association for male breast cancer (relative risk for ≥1-year of use before breast cancer diagnosis compared with <1-year of use: 0.70: 95% CI 0.34, 1.45). In the second study, the estimated odds ratio for breast cancer associated with the use of 5-ARIs compared with non-use was 1.08: 95% CI 0.62, 1.87).
The relationship between long term use of dutasteride and male breast cancer has not been established.
Pharmacokinetics: Absorption: Dutasteride is administered orally in solution as a soft gelatin capsule. Following administration of a single 0.5 mg dose, peak serum concentrations of dutasteride occur within 1 to 3 hours.
Absolute bioavailability in man is approximately 60% relative to a 2 hours intravenous infusion. The bioavailability of dutasteride is not affected by food.
Distribution: Pharmacokinetic data following single and repeat oral doses show that dutasteride has a large volume of distribution (300 to 500 L). Dutasteride is highly bound to plasma proteins (>99.5%).
Following daily dosing, dutasteride serum concentrations achieve 65% of steady state concentration after 1 month and approximately 90% after 3 months. Steady state serum concentrations (Css) of approximately 40 nanograms/mL are achieved after 6 months of dosing 0.5 mg once a day. Similarly to serum, dutasteride concentrations in semen achieved steady state at 6 months. After 52 weeks of therapy, semen dutasteride concentrations averaged 3.4 nanograms/mL (range 0.4 to 14 nanograms/mL). Dutasteride partitioning from serum into semen averaged 11.5%.
Biotransformation: In vitro, dutasteride is metabolized by the human cytochrome P450 isoenzyme CYP3A4 to two minor monohydroxylated metabolites, but it is not metabolized by CYP1A2, CYP2A6, CYP2E1, CYP2C8, CYP2C9, CYP2C19, CYP2B6, or CYP2D6.
In human serum, following dosing to steady state, unchanged dutasteride, 3 major metabolites (4'-hydroxydutasteride; 1,2-dihydrodutasteride and 6-hydroxydutasteride) and 2 minor metabolites (6,4'-dihydroxydutasteride and 15-hydroxydutasteride), as assessed by mass spectrometric response, have been detected. The five human serum metabolites of dutasteride have been detected in rat serum, however the stereochemistry of the hydroxyl additions at the 6 and 15 positions in the human and rat metabolites is not known.
Elimination: Dutasteride is extensively metabolized. Following oral dosing of dutasteride 0.5 mg/day to steady state in humans, 1.0% to 15.4% (mean of 5.4%) of the administered dose is excreted as dutasteride in the faeces. The remainder is excreted in the faeces as 4 major metabolites comprising 39%, 21%, 7% and 7% each of drug-related material and 6 minor metabolites (less than 5% each).
Only trace amounts of unchanged dutasteride (less than 0.1% of the dose) are detected in human urine.
At therapeutic concentrations, the terminal half-life of dutasteride is 3 to 5 weeks.
Serum concentrations remain detectable (greater than 0.1 nanograms/mL) for up to 4 to 6 months after discontinuation of treatment.
Linearity/Nonlinearity: Dutasteride pharmacokinetics can be described as first order absorption process and two parallel elimination pathways, one saturable (concentration-dependent) and one non-saturable (concentration-independent).
At low serum concentrations (less than 3 nanograms/mL), dutasteride is cleared rapidly by both the concentration-dependent and concentration-independent elimination pathways. Single doses of 5 mg or less showed evidence of rapid clearance and a short half-life of 3 to 9 days.
At serum concentrations, greater than 3 nanograms/mL, dutasteride is cleared slowly (0.35 to 0.58 L/h) primarily by linear, non-saturable elimination with terminal half-life of 3 to 5 weeks. At therapeutic concentrations, following repeat dosing of 0.5 mg/day, the slower clearance dominates and the total clearance is linear and concentration-independent.
Elderly: Dutasteride pharmacokinetics and pharmacodynamics were evaluated in 36 healthy male subjects between the ages of 24 and 87 years following administration of a single 5 mg dose of dutasteride. Exposure of dutasteride, represented by AUC and Cmax values, was not statistically different when comparing age groups. Half-life was not statistically different when comparing the 50 to 69 years old group to the greater than 70 years old group, which encompasses the age of most men with BPH. No differences in drug effect as measured by DHT reduction were observed between age groups. Results indicated that no dutasteride dose-adjustment based on age is necessary.
Renal Impairment: The effect of renal impairment on dutasteride pharmacokinetics has not been studied. However, less than 0.1% of a steady state 0.5 mg dose of dutasteride is recovered in human urine, so no adjustment in dosage is anticipated for patients with renal impairment.
Hepatic Impairment: The effect on the pharmacokinetics of dutasteride in hepatic impairment has not been studied. (See Precautions.)
Toxicology: Preclinical Safety Data: At exposures greatly in excess of those at the clinical dose, reversible, nonspecific CNS-related effects were seen in rats (425-fold) and dogs (315-fold).
Other toxicity findings were consistent with the pharmacological activity of 5-alpha-reductase inhibition. In male rats and dogs, these included effects on accessory reproductive organs and, in male rats, a reversible decrease in fertility. This is considered to have no clinical relevance as there was no effect on sperm development, concentration or motility. Feminisation of the external genitalia was noted in male foetuses of female rats and rabbits orally dosed with dutasteride. However, intravenous administration of dutasteride to pregnant Rhesus monkeys during embryofetal development at doses of up to 2,010 nanograms/animal/day did not produce adverse maternal or foetal toxicity. This dose represents a multiple of at least 186-fold (nanograms/kg basis) the potential maximum daily dose in a 50 kg woman, resulting from exposure to 5 mL semen (assuming 100% absorption) from a dutasteride-treated man.
Dutasteride was not genotoxic in a wide range of mutagenicity tests.
In a carcinogenicity study in rats, there was an increase in benign interstitial cell tumours in the testis at the high dose (158-fold clinical exposure). However, the endocrine mechanisms believed to be involved in the production of interstitial cell hyperplasia and adenomas in the rat are not relevant to humans. There were no clinically relevant effects on tumour profile in a carcinogenicity study in mice.
Avodart treats and prevents progression of benign prostatic hyperplasia (BPH) through alleviating symptoms, reducing prostate size (volume), improving urinary flow rate and reducing the risk of acute urinary retention (AUR) and the need for BPH-related surgery.
Avodart in combination with alpha-blocker tamsulosin, is indicated for the treatment of moderate to severe symptomatic benign prostatic hyperplasia (BPH) in men with enlarged prostate.
Avodart can be administered alone or in combination with the alpha-blocker tamsulosin (0.4 mg).
The recommended dose of Avodart is one capsule (0.5 mg) taken orally once a day.
The capsules should be swallowed whole and not chewed or opened, as contact with the capsule contents may result in irritation of the oropharyngeal mucosa. The capsules may be taken with or without food. Although an improvement may be observed at an early stage, it can take up to 6 months before a response to the treatment can be achieved. No dose adjustment is necessary in the elderly.
Renal Impairment: The effect of renal impairment on dutasteride pharmacokinetics has not been studied. However, no adjustment in dosage is anticipated for patients with renal impairment (see Pharmacokinetics under Actions).
Hepatic Impairment: The effect of hepatic impairment on dutasteride pharmacokinetics has not been studied so caution should be used in patients with mild to moderate hepatic impairment (see Pharmacokinetics under Actions and Precautions). In patients with hepatic impairment, the use of dutasteride is contraindicated.
In volunteer studies single doses of dutasteride up to 40 mg/day (80 times the therapeutic dose) for 7 days have been administered without significant safety concerns. In clinical studies doses of 5 mg daily have been administered to patients for 6 months with no additional adverse effects to those seen at therapeutic doses of 0.5 mg.
There is no specific antidote for dutasteride; therefore, in cases of suspected overdosage, symptomatic and supportive treatment should be given as appropriate.
Avodart is contraindicated in women and children and adolescents (see Use in Pregnancy & Lactation). Patients with hypersensitivity to dutasteride, other 5-alpha-reductase inhibitors, or any of the excipients (see Description). Patients with severe hepatic impairment.
Combination therapy should be prescribed after careful benefit risk assessment due to the potential increased risk of adverse events and after consideration of alternative treatment options including monotherapies (see Dosage & Administration).
Prostate cancer: In a 4-year study of over 8,000 men aged 50 to 75, with a prior negative biopsy for prostate cancer and baseline PSA between 2.5 nanograms/mL and 10.0 nanograms/mL (the REDUCE study), 1,517 men were diagnosed with prostate cancer. There was a higher incidence of Gleason 8-10 prostate cancers in the Avodart group (n=29, 0.9%) compared to the placebo group (n=19, 0.6%). There was no increased incidence in Gleason 5-6 or 7-10 prostate cancers. No causal relationship between Avodart and high-grade prostate cancer has been established. The clinical significance of the numerical imbalance is unknown. Men taking Avodart should be regularly evaluated for prostate cancer risk including PSA testing.
In an additional 2-year follow-up study with the original patients from the dutasteride chemoprevention study (REDUCE), a low rate of new prostate cancers were diagnosed (dutasteride [n=14, 1.2%] and placebo [n=7, 0.7%]), with no new identified cases of Gleason 8-10 prostate cancers.
Long-term follow up (up to 18 years) of another 5-ARI (finasteride) in a chemoprevention study showed no statistically significant difference between finasteride and placebo in the rates of overall survival (HR 1.02, 95% CI 0.97-1.08) or survival after prostate cancer diagnoses (HR 1.01, 95% CI 0.85-1.20).
Prostate specific antigen (PSA): Dutasteride was not studied in patients with liver disease. Caution should be used in the administration of dutasteride to patients with mild to moderate hepatic impairment (see Dosage & Administration, Contraindications, and Pharmacokinetics under Actions).
Serum prostate-specific antigen (PSA) concentration is an important component of the screening process to detect prostate cancer.
Avodart causes a decrease in mean serum PSA levels by approximately 50% after 6 months of treatment.
Patients receiving Avodart should have a new PSA baseline established after 6 months of treatment with Avodart. It is recommended to monitor PSA values regularly thereafter. Any confirmed increase from lowest PSA level while on Avodart may signal the presence of prostate cancer or non-compliance to therapy with Avodart and should be carefully evaluated, even if those values are still within the normal range for men not taking a 5-ARI. In the interpretation of a PSA value for a patient taking Avodart, previous PSA values should be sought for comparison.
Treatment with Avodart does not interfere with the use of PSA as a tool to assist in the diagnosis of prostate cancer after a new baseline has been established.
Total serum PSA levels return to baseline within 6 months of discontinuing treatment.
The ratio of free to total PSA remains constant even under the influence of Avodart. If clinicians elect to use percent-free PSA as an aid in the detection of prostate cancer in men undergoing Avodart therapy, no adjustment to its value is necessary.
Digital rectal examination, as well as other evaluations for prostate cancer, should be performed on patients prior to initiating therapy with Avodart and periodically thereafter.
Cardiovascular adverse events: In two 4-year clinical studies, the incidence of cardiac failure (a composite term of reported events, primarily cardiac failure and congestive cardiac failure) was higher among subjects taking the combination of Avodart and an alpha-blocker, primarily tamsulosin, than it was among subjects not taking the combination. In these two trials, the incidence of cardiac failure was low (≤1%) and variable between the studies. No imbalance was observed in the incidence of cardiovascular adverse events overall in either trial. No causal relationship between Avodart (alone or in combination with an alpha blocker) and cardiac failure has been established (see Clinical Studies under Actions).
In a meta-analysis of 12-randomised, placebo- or comparator-controlled clinical studies (n=18,802) that evaluated the risks of developing cardiovascular adverse events from the use of Avodart (by comparison with controls), no consistent statistically significant increase in the risk of heart failure (RR 1.05; 95% CI 0.71, 1.57), acute myocardial infarction (RR 1.00; 95% CI 0.77, 1.30) or stroke (RR 1.20; 95% CI 0.88, 1.64) were found.
Breast cancer: There have been rare reports of male breast cancer reported in men taking Avodart in clinical trials and during the post-marketing period. However, epidemiological studies showed no increase in the risk of developing male breast cancer with the use of 5-ARIs (see Clinical Studies under Actions). Prescribers should instruct their patients to promptly report any changes in their breast tissue such as lumps or nipple discharge.
Leaking capsules: Dutasteride is absorbed through the skin, therefore women and children must avoid contact with leaking capsules (see Use in Pregnancy & Lactation). If contact is made with leaking capsules the contact area should be washed immediately with soap and water.
Effects on Ability to Drive and Use Machines: Based on the pharmacokinetic and pharmacodynamic properties of dutasteride, treatment would not be expected to interfere with the ability to drive or operate machinery.
Hepatic impairment: The effect of hepatic impairment on dutasteride pharmacokinetics has not been studied. Because dutasteride is extensively metabolised and has a half-life of 3 to 5 weeks, caution should be used in the administration of dutasteride to patients with liver disease (see Dosage & Administration and Pharmacokinetics under Actions).
Fertility: The effects of dutasteride 0.5 mg/day on semen characteristics were evaluated in normal volunteers aged 18 to 52 (n=27 dutasteride, n=23 placebo) throughout 52 weeks of treatment and 24 weeks of post treatment follow-up. At 52 weeks, the mean percent reduction from baseline in total sperm count, semen volume, and sperm motility were 23%, 26%, and 18%, respectively, in the dutasteride group when adjusted for changes from baseline in the placebo group. Sperm concentration and sperm morphology were unaffected. After 24 weeks of follow-up, the mean percent change in total sperm count in the dutasteride group remained 23% lower than baseline. While mean values for all semen parameters at all time points remained within the normal ranges and did not meet predefined criteria for a clinically significant change (30%), two subjects in the dutasteride group had decreases in sperm count of greater than 90% from baseline at 52 weeks, with partial recovery at the 24-week follow-up. The clinical significance of dutasteride's effect on semen characteristics for an individual patient’s fertility is not known.
Pregnancy: Dutasteride is contraindicated for use by women. Dutasteride has not been studied in women because pre-clinical data suggests that the suppression of circulating levels of dihydrotestosterone may inhibit the development of the external genital organs in a male foetus carried by a woman exposed to dutasteride.
Lactation: It is not known whether dutasteride is excreted in breast milk.
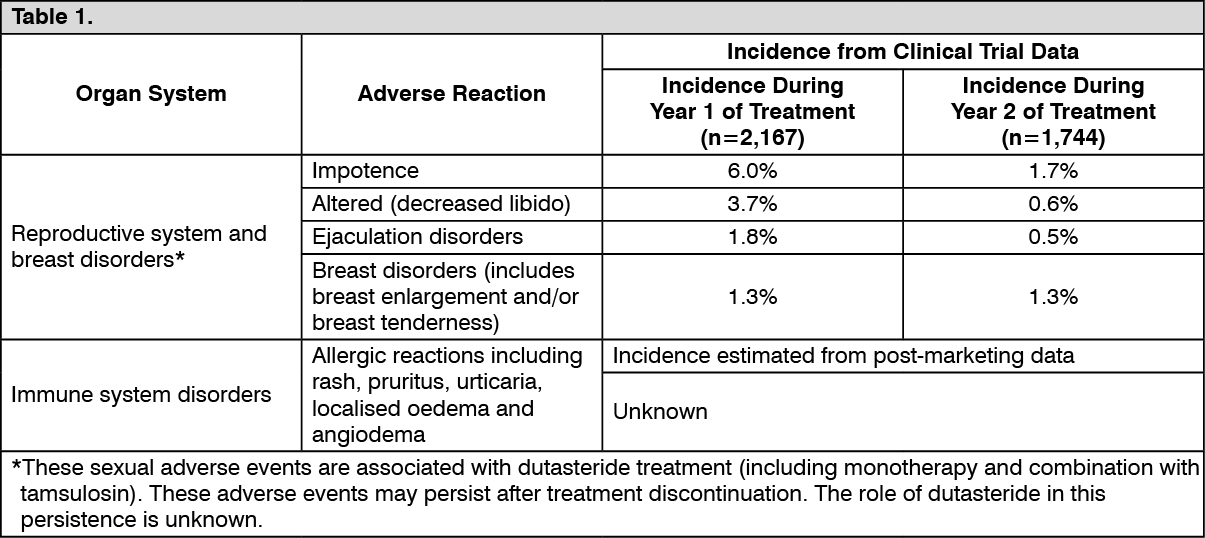
Clinical Trial Data: Avodart Monotherapy for BPH: Approximately 19% of the 2,167 patients who received dutasteride in the 2-year Phase III placebo-controlled trials developed adverse reactions during the first year of treatment. The majority of events were mild to moderate and occurred in the reproductive system. No change to the adverse event profile was apparent over a further 2 year in open-label extension studies.
Table 1 shows adverse reactions from controlled clinical trials and post-marketing experience. The listed adverse events from clinical trials are investigator-judged drug-related events (with incidence more than or equal to 1%) reported with a higher incidence in patients treated with dutasteride compared with placebo during the first year of treatment. Adverse events from post-marketing experience were identified from spontaneous post-marketing reports, therefore, the true incidence is unknown. (See Table 1.)
 Click on icon to see table/diagram/image
Avodart and Tamsulosin Combination Therapy for BPH:
Click on icon to see table/diagram/image
Avodart and Tamsulosin Combination Therapy for BPH: The following investigator-judged drug-related adverse events (with a cumulative incidence of greater than or equal to 1%) have been reported in the CombAT (Combination of Avodart and Tamsulosin) study, a comparison of Avodart 0.5 mg and tamsulosin 0.4 mg once daily for four years in combination or as monotherapy. (See Table 2.)
Click on icon to see table/diagram/image
Post-Marketing Data: Adverse drug reactions are listed by system organ class and frequency.
Frequencies are defined as: very common (≥1/10), common (≥1/100 to <1/10), uncommon (≥1/1,000 to <1/100), rare (≥1/10,000 to <1/1,000) and very rare (<1/10,000) including isolated reports. Frequency categories determined from post-marketing data refer to reporting rate rather than true frequency.
Immune System Disorders: Very Rare: Allergic reactions, including rash, pruritus, urticaria, localised oedema and angioedema.
Psychiatric Disorders: Very Rare: Depressed mood.
Skin and Subcutaneous Tissue Disorders: Rare: Alopecia (primary body hair loss), hypertrichosis.
Reproductive System and Breast Disorders: Very Rare: Testicular pain and testicular swelling.
For information on the decrease of serum PSA levels during treatment with dutasteride and guidance concerning prostate cancer detection see Precautions.
Effects of Other Drugs on the Pharmacokinetics of Dutasteride: Use Together with CYP3A4 and/or P-Glycoprotein-Inhibitors: Dutasteride is mainly eliminated via metabolism. In vitro studies indicate that this metabolism is catalysed by CYP3A4 and CYP3A5. No formal interaction studies have been performed with potent CYP3A4 inhibitors. However, in a population pharmacokinetic study, dutasteride serum concentration were on average 1.6-1.8 times greater, respectively, in a small number of patients treated concurrently with verapamil or diltiazem (moderate inhibitors of CYP3A4 and inhibitors of P-glycoprotein) than in other patients.
Long-term combination of dutasteride with drugs that are potent inhibitors of the enzyme CYP3A4 (eg, ritonavir, indinavir, nefazodone, itraconazole, ketoconazole administered orally) may increase serum concentrations of dutasteride. Further inhibition of 5-alpha-reductase at increased dutasteride exposure, is not likely. However, a reduction of the dutasteride dosing frequency can be considered if side effects are noted. It should be noted that in the case of enzyme inhibition, the long half-life may be further prolonged and it can take more than 6 months of concurrent therapy before a new steady-state is reached.
Administration of 12 g colestyramine one hour before a 5 mg single dose of dutasteride did not affect the pharmacokinetics of dutasteride.
Effects of Dutasteride on the Pharmacokinetics of Other Drugs: Dutasteride has no effect on the pharmacokinetics of warfarin or digoxin. This indicates that dutasteride does not inhibit/induce CYP2C9 or the transporter P-glycoprotein. In vitro interaction studies indicate that dutasteride does not inhibit the enzyme CYP1A2, CYP2D6, CYP2C9, CYP2C19 or CYP3A4.
In a small study (N=24) of two weeks duration in healthy men, dutasteride (0.5 mg daily) had no effect on the pharmacokinetics of tamsulosin or terazosin. There was also no indication of a pharmacodynamic interaction in this study.
Instructions for Use/Handling: Dutasteride is absorbed through the skin; therefore, women and children must avoid contact with leaking capsules (see Precautions and Use in Pregnancy & Lactation). If contact is made with leaking capsules, the contact area should be washed immediately with soap and water.
Incompatibilities: Not applicable.
G04CB02 - dutasteride ; Belongs to the class of testosterone-5-alpha reductase inhibitors. Used in the treatment of benign prostatic hypertrophy.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image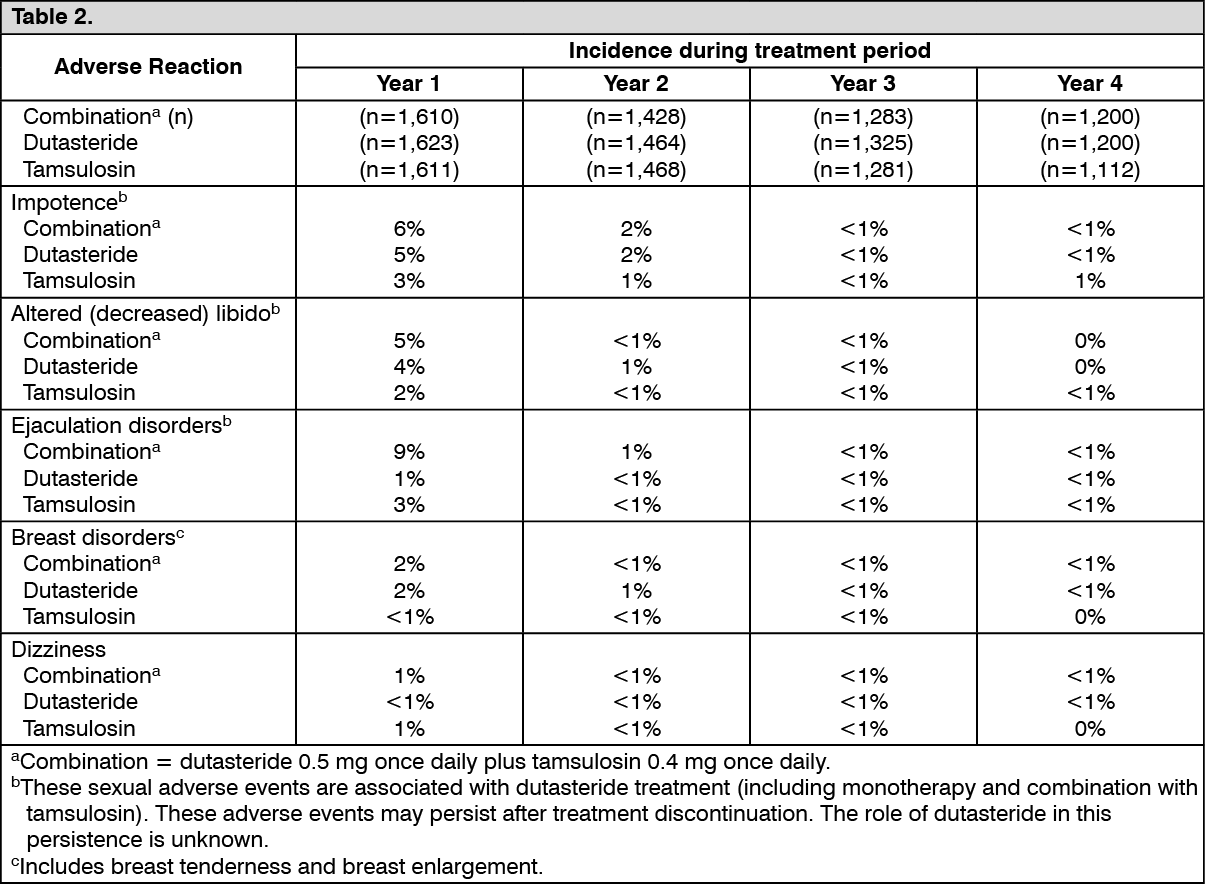 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out