Pharmacotherapeutic group: Drugs for the treatment of bone diseases - Other drugs affecting bone structure and mineralisation.
ATC code: M05BX04.
Pharmacology: Pharmacodynamics: Mechanism of action: Denosumab is a human monoclonal antibody (IgG2) that targets and binds with high affinity and specificity to RANKL preventing RANKL from activating its only receptor, RANK, on the surface of osteoclasts and their precursors, independent of bone surface. Prevention of the RANKL/RANK interaction inhibits osteoclast formation, function, and survival. Denosumab therefore reduces bone resorption and increases bone mass and strength in both cortical and trabecular bone.
Pharmacodynamic effects: At the end of each dosing interval, CTX reductions were partially attenuated from maximal reduction of ≥ 87% to approximately ≥ 45% (range 45-80%), reflecting the reversibility of denosumab's effects on bone remodelling once serum levels diminish. These effects were sustained with continued treatment. Bone turnover markers generally reached pre-treatment levels within 9 months after the last dose. Upon re-initiation, the degree of inhibition of CTX by denosumab was similar to that observed in patients initiating denosumab treatment.
Immunogenicity: In clinical studies, neutralising antibodies have not been observed for Prolia. Using a sensitive immunoassay < 1% of patients treated with denosumab for up to 5 years tested positive for non neutralising binding antibodies with no evidence of altered pharmacokinetics, toxicity, or clinical response.
Clinical efficacy and safety in postmenopausal women with osteoporosis: Efficacy and safety of Prolia administered once every 6 months for 3 years were investigated in post-menopausal women (7,808 women aged 60-91 years, of which 23.6% had prevalent vertebral fractures) with baseline bone mineral density (BMD) T-scores at the lumbar spine or total hip between -2.5 and -4.0 and a mean absolute 10-year fracture probability of 18.60% (deciles: 7.9-32.4%) for major osteoporotic fracture and 7.22% (deciles: 1.4-14.9%) for hip fracture. Women with other diseases or on therapies that may affect bone were excluded from this study. Women received calcium (at least 1,000 mg) and vitamin D (at least 400 IU) supplementation daily.
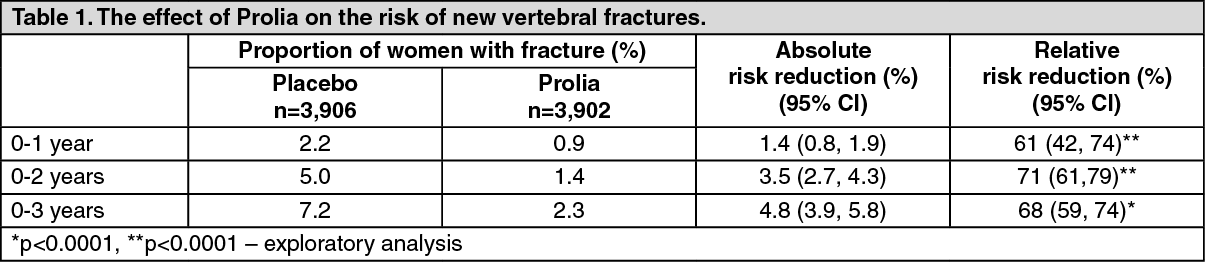
Effect on vertebral fractures: Prolia significantly reduced the risk of new vertebral fractures at 1, 2 and 3 years (p <0.0001) (see Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Effect on hip fractures: Prolia demonstrated a 40% relative reduction (0.5% absolute risk reduction) in the risk of hip fracture over 3 years (p <0.05). The incidence of hip fracture was 1.2% in the placebo group compared to 0.7% in the Prolia group at 3 years.
In a post-hoc analysis in women >75 years, a 62% relative risk reduction was observed with Prolia (1.4% absolute risk reduction, p<0.01).
Effect on all clinical fractures: Prolia significantly reduced fractures across all fracture types/groups (see Table 2.)
Click on icon to see table/diagram/image
In women with baseline femoral neck BMD ≤ -2.5, Prolia reduced the risk of non-vertebral fracture (35% relative risk reduction, 4.1% absolute risk reduction, p < 0.001, exploratory analysis).
The reduction in the incidence of new vertebral fractures, hip fractures and non-vertebral fractures by Prolia over 3 years were consistent regardless of the 10-year baseline fracture risk.
Effect on bone mineral density: Prolia significantly increased BMD at all clinical sites measured, versus placebo at 1, 2 and 3 years. Prolia increased BMD by 9.2% at the lumbar spine, 6.0% at the total hip, 4.8% at the femoral neck, 7.9% at the hip trochanter, 3.5% at the distal 1/3 radius and 4.1% at the total body over 3 years (all p < 0.0001).
In clinical studies examining the effects of discontinuation of Prolia, BMD returned to approximately pre-treatment levels and remained above placebo within 18 months of the last dose. These data indicate that continued treatment with Prolia is required to maintain the effect of the medicinal product. Re-initiation of Prolia resulted in gains in BMD similar to those when Prolia was first administered.
Open-label extension study in the treatment of postmenopausal osteoporosis A total of 4,550 women (2,343 Prolia & 2,207 placebo) who missed no more than one dose of investigational product in the pivotal study described above and completed the month 36 study visit agreed to enrol in a 7-year, multinational, multicentre, open-label, single-arm extension study to evaluate the long-term safety and efficacy of Prolia. All women in the extension study were to receive Prolia 60 mg every 6 months, as well as daily calcium (at least 1 g) and vitamin D (at least 400 IU). A total of 2,626 subjects (58% of the women included in the extension study i.e. 34% of the women included in the pivotal study) completed the extension study.
In patients treated with Prolia for up to 10 years, BMD increased from the pivotal study baseline by 21.7% at the lumbar spine, 9.2% at the total hip, 9.0% at the femoral neck, 13.0% at the trochanter and 2.8% at the distal 1/3 radius. The mean lumbar spine BMD T-score at the end of the study was −1.3 in patients treated for 10 years.
Fracture incidence was evaluated as a safety endpoint. but efficacy in fracture prevention cannot be estimated due to high number of discontinuations and open-label design. The cumulative incidence of new vertebral and non-vertebral fractures were approximately 6.8% and 13.1% respectively, in patients who remained on denosumab treatment for 10 years (n = 1,278). Patients who did not complete the study for any reason had higher on-treatment fracture rates.
Thirteen adjudicated cases of osteonecrosis of the jaw (ONJ) and two adjudicated cases of atypical fractures of the femur occurred during the extension study.
Clinical efficacy and safety in men with osteoporosis: Efficacy and safety of Prolia once every 6 months for 1 year were investigated in 242 men aged 31-84 years. Subjects with an eGFR < 30 mL/min/1.73 m
2 were excluded from the study. All men received calcium (at least 1,000 mg) and vitamin D (at least 800 IU) supplementation daily.
The primary efficacy variable was percent change in lumbar spine BMD, fracture efficacy was not evaluated. Prolia significantly increased BMD at all clinical sites measured, relative to placebo at 12 months: 4.8% at lumbar spine, 2.0% at total hip, 2.2% at femoral neck, 2.3% at hip trochanter, and 0.9% at distal 1/3 radius (all p < 0.05). Prolia increased lumbar spine BMD from baseline in 94.7% of men at 1 year. Significant increases in BMD at lumbar spine, total hip, femoral neck and hip trochanter were observed by 6 months (p < 0.0001).
Bone histology in postmenopausal women and men with osteoporosis: Bone histology was evaluated in 62 postmenopausal women with osteoporosis or with low bone mass who were either naïve to osteoporosis therapies or had transitioned from previous alendronate therapy following 1-3 years treatment with Prolia. Fifty nine women participated in the bone biopsy sub-study at month 24 (n = 41) and/or month 84 (n = 22) of the extension study in postmenopausal women with osteoporosis. Bone histology was also evaluated in 17 men with osteoporosis following 1 year treatment with Prolia. Bone biopsy results showed bone of normal architecture and quality with no evidence of mineralisation defects, woven bone or marrow fibrosis. Histomorphometry findings in the extension study in postmenopausal women with osteoporosis showed that the antiresorptive effects of Prolia, as measured by activation frequency and bone formation rates, were maintained over time.
Clinical efficacy and safety in patients with bone loss associated with androgen deprivation: Efficacy and safety of Prolia once every 6 months for 3 years were investigated in men with histologically confirmed non-metastatic prostate cancer receiving ADT (1,468 men aged 48-97 years) who were at increased risk of fracture (defined as > 70 years, or < 70 years with a BMD T-score at the lumbar spine, total hip, or femoral neck < -1.0 or a history of an osteoporotic fracture.) All men received calcium (at least 1,000 mg) and vitamin D (at least 400 IU) supplementation daily.
Prolia significantly increased BMD at all clinical sites measured, relative to treatment with placebo at 3 years: 7.9% at the lumbar spine, 5.7% at the total hip, 4.9% at the femoral neck, 6.9% at the hip trochanter, 6.9% at the distal 1/3 radius and 4.7% at the total body (all p < 0.0001). In a prospectively planned exploratory analysis, significant increases in BMD were observed at the lumbar spine, total hip, femoral neck and the hip trochanter 1 month after the initial dose.
Prolia demonstrated a significant relative risk reduction of new vertebral fractures: 85% (1.6% absolute risk reduction) at 1 year, 69% (2.2% absolute risk reduction) at 2 years and 62% (2.4% absolute risk reduction) at 3 years (all p < 0.01).
Clinical efficacy and safety in patients with bone loss associated with adjuvant aromatase inhibitor therapy: Efficacy and safety of Prolia once every 6 months for 2 years were investigated in women with non-metastatic breast cancer (252 women aged 35-84 years) and baseline BMD T-scores between -1.0 to -2.5 at the lumbar spine, total hip or femoral neck. All women received calcium (at least 1,000 mg) and vitamin D (at least 400 IU) supplementation daily.
The primary efficacy variable was percent change in lumbar spine BMD, fracture efficacy was not evaluated. Prolia significantly increased BMD at all clinical sites measured, relative to treatment with placebo at 2 years: 7.6% at lumbar spine, 4.7% at total hip, 3.6% at femoral neck, 5.9% at hip trochanter, 6.1% at distal 1/3 radius and 4.2% at total body (all p < 0.0001).
Treatment of bone loss associated with long-term systemic glucocorticoid therapy: Efficacy and safety of Prolia were investigated in 795 patients (70% women and 30% men) aged 20 to 94 years treated with ≥ 7.5 mg daily oral prednisone (or equivalent).
Two subpopulations were studied: glucocorticoid-continuing (≥ 7.5 mg daily prednisone or its equivalent for ≥ 3 months prior to study enrolment; n = 505) and glucocorticoid-initiating (≥ 7.5 mg daily prednisone or its equivalent for < 3 months prior to study enrolment; n = 290). Patients were randomised (1:1) to receive either Prolia 60 mg subcutaneously once every 6 months or oral risedronate 5 mg once daily (active control) for 2 years. Patients received calcium (at least 1,000 mg) and vitamin D (at least 800 IU) supplementation daily.
Effect on Bone Mineral Density (BMD): In the glucocorticoid-continuing subpopulation, Prolia demonstrated a greater increase in lumbar spine BMD compared to risedronate at 1 year (Prolia 3.6%, risedronate 2.0%; p < 0.001) and 2 years (Prolia 4.5%, risedronate 2.2%; p < 0.001). In the glucocorticoid-initiating subpopulation, Prolia demonstrated a greater increase in lumbar spine BMD compared to risedronate at 1 year (Prolia 3.1%, risedronate 0.8%; p < 0.001) and 2 years (Prolia 4.6%, risedronate 1.5%; p < 0.001).
In addition, Prolia demonstrated a significantly greater mean percent increase in BMD from baseline compared to risedronate at the total hip, femoral neck, and hip trochanter.
The study was not powered to show a difference in fractures. At 1 year, the subject incidence of new radiological vertebral fracture was 2.7% (denosumab) versus 3.2% (risedronate). The subject incidence of non-vertebral fracture was 4.3% (denosumab) versus 2.5% (risedronate). At 2 years, the corresponding numbers were 4.1% versus 5.8% for new radiological vertebral fractures and 5.3% versus 3.8% for non-vertebral fractures. Most of the fractures occurred in the GC-C subpopulation.
Pharmacokinetics: Absorption: Following subcutaneous administration of a 1.0 mg/kg dose, which approximates the approved 60 mg dose, exposure based on AUC was 78% as compared to intravenous administration at the same dose level. For a 60 mg subcutaneous dose, maximum serum denosumab concentrations (C
max) of 6 μg/mL (range 1-17 μg/mL) occurred in 10 days (range 2-28 days).
Biotransformation: Denosumab is composed solely of amino acids and carbohydrates as native immunoglobulin and is unlikely to be eliminated via hepatic metabolic mechanisms. Its metabolism and elimination are expected to follow the immunoglobulin clearance pathways, resulting in degradation to small peptides and individual amino acids.
Elimination: After C
max, serum levels declined with a half-life of 26 days (range 6-52 days) over a period of 3 months (range 1.5-4.5 months). Fifty-three percent (53%) of patients had no measurable amounts of denosumab detected at 6 months post-dose.
No accumulation or change in denosumab pharmacokinetics with time was observed upon subcutaneous multiple-dosing of 60 mg once every 6 months. Denosumab pharmacokinetics were not affected by the formation of binding antibodies to denosumab and were similar in men and women. Age (28-87 years), race and disease state (low bone mass or osteoporosis; prostate or breast cancer) do not appear to significantly affect the pharmacokinetics of denosumab.
A trend was observed between higher body weight and lower exposure based on AUC and C
max. However, the trend is not considered clinically important, since pharmacodynamic effects based on bone turnover markers and BMD increases were consistent across a wide range of body weight.
Linearity/non-linearity: In dose ranging studies, denosumab exhibited non-linear, dose-dependent pharmacokinetics, with lower clearance at higher doses or concentrations, but approximately dose-proportional increases in exposures for doses of 60 mg and greater.
Renal impairment: In a study of 55 patients with varying degrees of renal function, including patients on dialysis, the degree of renal impairment had no effect on the pharmacokinetics of denosumab.
Hepatic impairment: No specific study in patients with hepatic impairment was performed. In general, monoclonal antibodies are not eliminated via hepatic metabolic mechanisms. The pharmacokinetics of denosumab is not expected to be affected by hepatic impairment.
Paediatric population: The pharmacokinetic profile in paediatric populations has not been assessed.
Toxicology: Preclinical safety data: In single and repeated dose toxicity studies in cynomolgus monkeys, denosumab doses resulting in 100 to 150 times greater systemic exposure than the recommended human dose had no impact on cardiovascular physiology, male or female fertility, or produced specific target organ toxicity.
Standard tests to investigate the genotoxicity potential of denosumab have not been evaluated, since such tests are not relevant for this molecule. However, due to its character it is unlikely that denosumab has any potential for genotoxicity.
The carcinogenic potential of denosumab has not been evaluated in long-term animal studies.In preclinical studies conducted in knockout mice lacking RANK or RANKL, impairment of lymph node formation was observed in the foetus. An absence of lactation due to inhibition of mammary gland maturation (lobulo-alveolar gland development during pregnancy) was also observed in knockout mice lacking RANK or RANKL.
In a study of cynomolgus monkeys dosed with denosumab during the period equivalent to the first trimester at AUC exposures up to 99-fold higher than the human dose (60 mg every 6 months), there was no evidence of maternal or foetal harm. In this study, foetal lymph nodes were not examined.
In another study of cynomolgus monkeys dosed with denosumab throughout pregnancy at AUC exposures 119-fold higher than the human dose (60 mg every 6 months), there were increased stillbirths and postnatal mortality; abnormal bone growth resulting in reduced bone strength, reduced haematopoiesis, and tooth malalignment; absence of peripheral lymph nodes; and decreased neonatal growth. A no observed adverse effect level for reproductive effects was not established. Following a 6 month period after birth, bone related changes showed recovery and there was no effect on tooth eruption. However, the effects on lymph nodes and tooth malalignment persisted, and minimal to moderate mineralisation in multiple tissues was seen in one animal (relation to treatment uncertain). There was no evidence of maternal harm prior to labour; adverse maternal effects occurred infrequently during labour. Maternal mammary gland development was normal.
In preclinical bone quality studies in monkeys on long-term denosumab treatment, decreases in bone turnover were associated with improvement in bone strength and normal bone histology. Calcium levels were transiently decreased and parathyroid hormone levels transiently increased in ovariectomised monkeys treated with denosumab.
In male mice genetically engineered to express huRANKL (knock-in mice), which were subjected to a transcortical fracture, denosumab delayed the removal of cartilage and remodelling of the fracture callus compared to control, but biomechanical strength was not adversely affected.
Knockout mice (see Use in Pregnancy & Lactation) lacking RANK or RANKL exhibited decreased body weight, reduced bone growth and lack of tooth eruption. In neonatal rats, inhibition of RANKL (target of denosumab therapy) with high doses of a construct of osteoprotegerin bound to Fc (OPG-Fc) was associated with inhibition of bone growth and tooth eruption. These changes were partially reversible in this model when dosing with RANKL inhibitors was discontinued. Adolescent primates dosed with denosumab at 27 and 150 times (10 and 50 mg/kg dose) the clinical exposure had abnormal growth plates. Therefore, treatment with denosumab may impair bone growth in children with open growth plates and may inhibit eruption of dentition.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image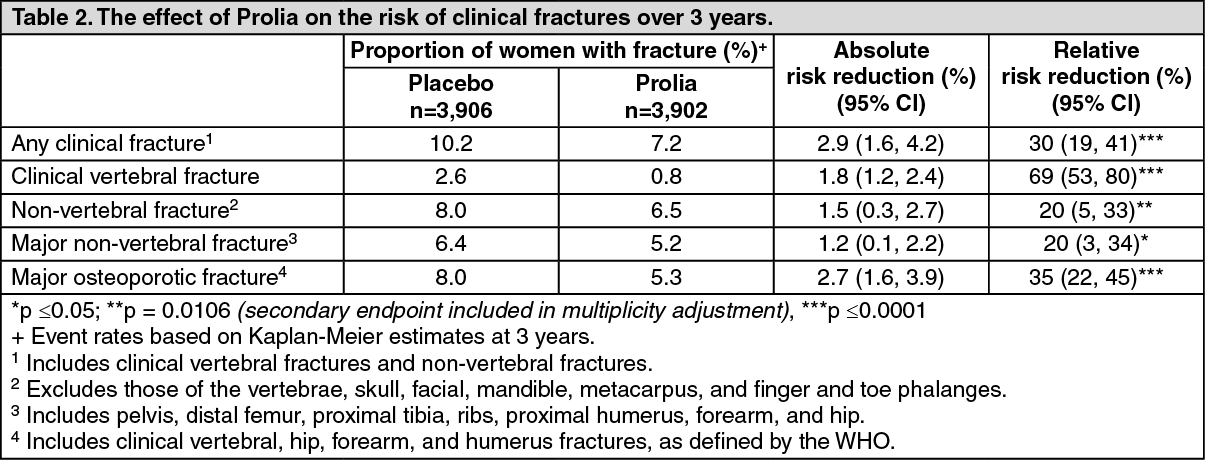 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image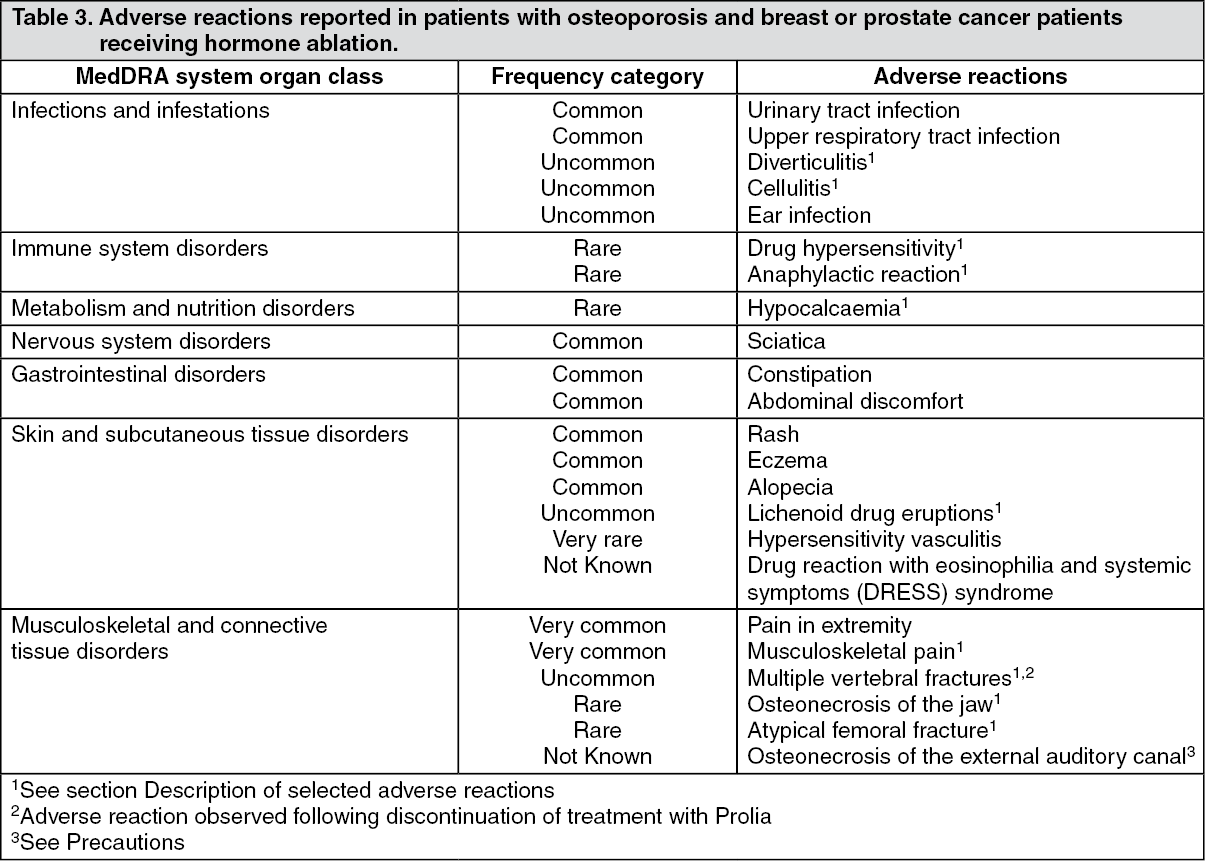 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image 60 mg_mLd35048de-8812-4dcf-9c96-a08600d5e04a.GIF)

 Sign Out
Sign Out
