Pharmacology: Pharmacodynamics: Sunitinib inhibits multiple receptor tyrosine kinases (RTKs) that are implicated in tumor growth, pathologic angiogenesis, and metastatic progression of cancer. Sunitinib was identified as an inhibitor of platelet-derived growth factor receptors (PDGFRα and PDGFRβ), VEGF receptors (VEGFR1, VEGFR2, and VEGFR3), stem cell factor receptor (KIT), Fms-like tyrosine kinase-3 (FLT3), colony stimulating factor receptor Type 1 (CSF-1R), and the glial cell-line derived neurotrophic factor receptor (RET). Sunitinib inhibition of the activity of these RTKs has been demonstrated in biochemical and cellular assays, and inhibition of function has been demonstrated in cell proliferation assays. The primary metabolite exhibits similar potency compared to sunitinib in biochemical and cellular assays.
Sunitinib inhibited the phosphorylation of multiple RTKs (PDGFRβ, VEGFR2, KIT) in tumor xenografts expressing RTK targets
in vivo and demonstrated inhibition of tumor growth or tumor regression, and/or inhibited in metastases in some experimental models of cancer. Sunitinib demonstrated the ability to inhibit growth of tumor cells expressing dysregulated target RTKs (PDGFR, RET, or KIT)
in vitro and to inhibit PDGFRβ- and VEGFR2-dependent tumor angiogenesis
in vivo.
The clinical safety and efficacy of sunitinib has been studied in subjects with malignant GIST who were resistant to imatinib (i.e., those who experienced disease progression during or following treatment with imatinib); or intolerant to imatinib (i.e., those who experienced significant toxicity during treatment with imatinib that precluded further treatment); in subjects with advanced renal cell carcinoma (RCC); and in subjects with unresectable pNET.
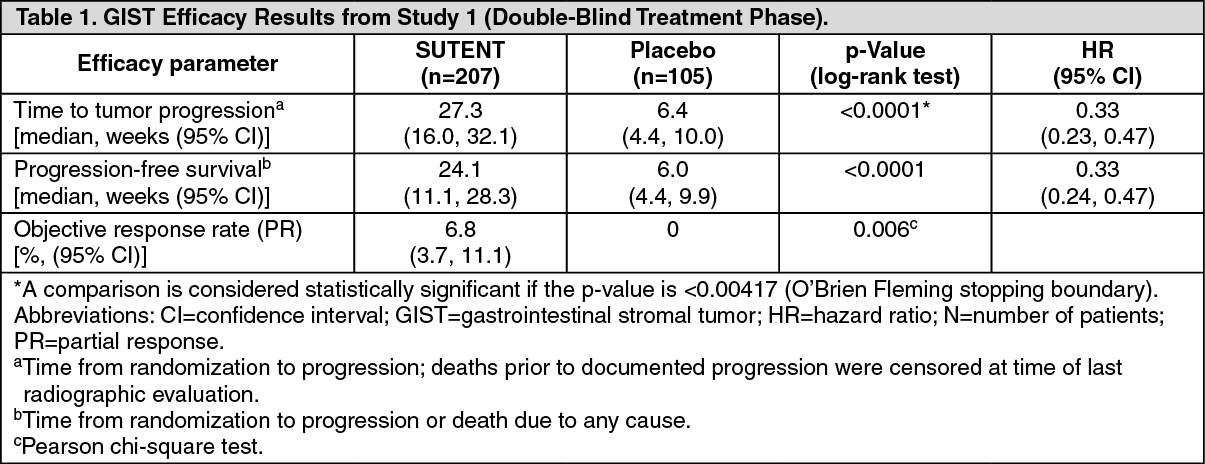
Efficacy is based on time to tumor progression and an increase in survival in GIST.
Efficacy is based on progression-free survival (PFS) and objective response rates (ORR) for treatment-naïve and cytokine-refractory advanced RCC, respectively and on PFS for pNET.
Clinical studies: Gastrointestinal Stromal Tumor: Study 1: Study 1 (NCT#00075218) was a 2-arm, international, randomized, double-blind, placebo-controlled trial of SUTENT in patients with GIST who had disease progression during prior imatinib mesylate (imatinib) treatment or who were intolerant of imatinib. The objective was to compare time-to-tumor progression (TTP) in patients receiving SUTENT plus best supportive care versus patients receiving placebo plus best supportive care. Other objectives included progression-free survival (PFS), objective response rate (ORR), and overall survival (OS). Patients were randomized (2:1) to receive either 50 mg SUTENT or placebo orally, once daily, on Schedule 4/2 until disease progression or withdrawal from the study for another reason. Treatment was unblinded at the time of disease progression. Patients randomized to placebo were then offered crossover to open-label SUTENT and patients randomized to SUTENT were permitted to continue treatment per investigator judgment.
At the time of a prespecified interim analysis, the intent-to-treat (ITT) population included 312 patients. Two hundred seven (207) patients were randomized to the SUTENT arm and 105 patients were randomized to the placebo arm. Demographics were comparable between the SUTENT and placebo groups with regard to age (69% versus 72% <65 years for SUTENT versus placebo, respectively), gender (male: 64% versus 61%), race (White: 88% both arms, Asian: 5% both arms, Black: 4% both arms, remainder not reported), and performance status (ECOG 0: 44% versus 46%, ECOG 1: 55% versus 52%, and ECOG 2: 1 versus 2%). Prior treatment included surgery (94% versus 93%) and radiotherapy (8% versus 15%). Outcome of prior imatinib treatment was also comparable between arms with intolerance (4% versus 4%), progression within 6 months of starting treatment (17% versus 16%), or progression beyond 6 months (78% versus 80%) balanced.
The planned interim efficacy and safety analysis was performed after 149 TTP events had occurred. There was a statistically significant advantage for SUTENT over placebo in TTP, meeting the primary endpoint. Efficacy results are summarized in Table 1 and the Kaplan-Meier curve for TTP is shown in Figure 1. (See Table 1 and Figure 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The final ITT population enrolled in the double-blind treatment phase of the study included 243 patients randomized to the SUTENT arm and 118 patients randomized to the placebo arm. After the primary endpoint was met at the interim analysis, the study was unblinded, and patients on the placebo arm were offered open-label SUTENT treatment. Ninety nine (99) of the patients initially randomized to placebo crossed over to receive SUTENT in the open-label treatment phase. At the protocol specified final analysis of OS, the median OS was 72.7 weeks for the SUTENT arm and 64.9 weeks for the placebo arm [hazard ratio (HR)=0.876, 95% confidence interval (CI) (0.679, 1.129)].
Study 2: Study 2 was an open-label, multi-center, single-arm, dose-escalation study conducted in patients with GIST following progression on or intolerance to imatinib. Following identification of the recommended regimen (50 mg once daily on Schedule 4/2), 55 patients in this study received the 50 mg dose of SUTENT on treatment Schedule 4/2. Partial responses (PR) were observed in 5 of 55 patients (9.1% PR rate, 95% CI 3.0%, 20.0%).
Renal Cell Carcinoma: Treatment-Naïve RCC: Study 3 (NCT#00083889) was a multi-center, international randomized study comparing single-agent SUTENT with IFN-α was conducted in patients with treatment-naïve RCC. The objective was to compare PFS in patients receiving SUTENT versus patients receiving IFN-α. Other endpoints included ORR, OS and safety. Seven hundred fifty (750) patients were randomized (1:1) to receive either 50 mg SUTENT once daily on Schedule 4/2 or to receive IFN-α administered subcutaneously at 9 million international units (MIU) 3 times a week. Patients were treated until disease progression or withdrawal from the study.
The ITT population included 750 patients, 375 randomized to SUTENT and 375 randomized to IFN-α. Demographics were comparable between the SUTENT and IFN-α groups with regard to age (59% versus 67% <65 years for SUTENT versus IFN-α, respectively), gender (Male: 71% versus 72%), race (White: 94% versus 91%, Asian: 2% versus 3%, Black: 1% versus 2%, remainder not reported), and performance status (ECOG 0: 62% versus 61%, ECOG 1: 38% each arm, ECOG 2: 0% versus 1%). Prior treatment included nephrectomy (91% versus 89%) and radiotherapy (14% each arm). The most common site of metastases present at screening was the lung (78% versus 80%, respectively), followed by the lymph nodes (58% versus 53%, respectively) and bone (30% each arm); the majority of the patients had multiple (2 or more) metastatic sites at baseline (80% versus 77%, respectively).
There was a statistically significant advantage for SUTENT over IFN-α in the endpoint of PFS (see Table 2 and Figure 2). In the prespecified stratification factors of lactate dehydrogenase (LDH) (>1.5 ULN versus ≤1.5 ULN), ECOG performance status (0 versus 1), and prior nephrectomy (yes versus no), the hazard ratio favored SUTENT over IFN-α. The ORR was higher in the SUTENT arm (see Table 2). (See Table 2 and Figure 2.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
At the protocol-specified final analysis of OS, the median OS was 114.6 weeks for the SUTENT arm and 94.9 weeks for the IFN-α arm (HR=0.821, 95% CI: 0.673, 1.001). The median OS for the IFN-α arm includes 25 patients who discontinued IFN-α treatment because of disease progression and crossed over to treatment with SUTENT as well as 121 patients (32%) on the IFN-α arm who received post-study cancer treatment with SUTENT.
Cytokine-Refractory RCC: The use of single-agent SUTENT in the treatment of cytokine-refractory RCC was investigated in 2 single-arm, multi-center studies. All patients enrolled into these studies experienced failure of prior cytokine-based therapy. In Study 4 (NCT#00077974), failure of prior cytokine therapy was based on radiographic evidence of disease progression defined by response evaluation criteria in solid tumors (RECIST) or World Health Organization (WHO) criteria during or within 9 months of completion of 1 cytokine therapy treatment (IFN-α, interleukin-2, or IFN-α plus interleukin-2; patients who were treated with IFN-α alone must have received treatment for at least 28 days). In Study 5 (NCT#00054886), failure of prior cytokine therapy was defined as disease progression or unacceptable treatment-related toxicity. The endpoint for both studies was ORR. Duration of response (DR) was also evaluated.
One hundred and six patients (106) were enrolled into Study 4 and 63 patients were enrolled into Study 5. Patients received 50 mg SUTENT on Schedule 4/2. Therapy was continued until the patients met withdrawal criteria or had progressive disease. The baseline age, gender, race and ECOG performance statuses of the patients were comparable between Studies 4 and 5. Approximately 86%-94% of patients in the 2 studies were White. Men comprised 65% of the pooled population. The median age was 57 years and ranged from 24 to 87 years in the studies. All patients had an ECOG performance status <2 at the screening visit.
The baseline malignancy and prior treatment history of the patients were comparable between Studies 4 and 5. Across the 2 studies, 95% of the pooled population of patients had at least some component of clear-cell histology. All patients in Study 4 were required to have a histological clear-cell component. Most patients enrolled in the studies (97% of the pooled population) had undergone nephrectomy; prior nephrectomy was required for patients enrolled in Study 4. All patients had received 1 previous cytokine regimen. Metastatic disease present at the time of study entry included lung metastases in 81% of patients. Liver metastases were more common in Study 4 (27% versus 16% in Study 5) and bone metastases were more common in Study 5 (51% versus 25% in Study 4); 52% of patients in the pooled population had at least 3 metastatic sites. Patients with known brain metastases or leptomeningeal disease were excluded from both studies.
The ORR and DR data from Studies 4 and 5 are provided in Table 3. There were 36 PRs in Study 4 as assessed by a core radiology laboratory for an ORR of 34.0% (95% CI 25.0%, 43.8%). There were 23 PRs in Study 5 as assessed by the investigators for an ORR of 36.5% (95% CI 24.7%, 49.6%). The majority (>90%) of objective disease responses were observed during the first 4 cycles; the latest reported response was observed in Cycle 10. DR data from Study 4 is premature as only 9 of 36 patients (25%) responding to treatment had experienced disease progression or died at the time of the data cutoff. (See Table 3.)
Click on icon to see table/diagram/image
Pancreatic Neuroendocrine Tumors: Study 6 (NCT#00428597) was a multi-center, international, randomized, double-blind placebo-controlled study of single-agent SUTENT conducted in patients with unresectable pNET. Patients were required to have documented RECIST-defined disease progression within the prior 12 months and were randomized (1:1) to receive either 37.5 mg SUTENT (n=86) or placebo (n=85) once daily without a scheduled off-treatment period. The primary objective was to compare PFS in patients receiving SUTENT versus patients receiving placebo. Other endpoints included OS, ORR, and safety. Use of somatostatin analogs was allowed in the study.
Demographics were comparable between the SUTENT and placebo groups. Additionally, 49% of SUTENT patients had non-functioning tumors vs. 52% of placebo patients, and 92% patients in both arms had liver metastases. A total of 66% of SUTENT patients received prior systemic therapy compared with 72% of placebo patients and 35% of SUTENT patients had received somatostatin analogs compared with 38% of placebo patients. Patients were treated until disease progression or withdrawal from the study. Upon disease progression or study closure, patients were offered access to SUTENT in a separate extension study.
As recommended by the Independent Data Monitoring Committee, the study was terminated prematurely prior to the pre-specified interim analysis. This may have led to an overestimate of the magnitude of PFS effect. A clinically significant improvement for SUTENT over placebo in PFS was seen by both investigator and independent assessment. A hazard ratio favoring SUTENT was observed in all subgroups of baseline characteristics evaluated. OS data were not mature at the time of the analysis. There were 9 deaths in the SUTENT arm and 21 deaths in the placebo arm. A statistically significant difference in ORR favoring SUTENT over placebo was observed. Efficacy results are summarized in Table 4 and the Kaplan-Meier curve for PFS is in Figure 3. (See Table 4 and Figure 3.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
A phase 4 multinational, multi-centre, single-arm, open-label study evaluating the efficacy and safety of sunitinib was conducted in patients with progressive, advanced/metastatic, well-differentiated, unresectable pNET.
One hundred six patients (61 patients in the treatment-naïve cohort and 45 patients in the later-line cohort) received treatment with sunitinib orally at 37.5 mg once a day on a continuous daily dosing (CDD) schedule.
The investigator-assessed median PFS was 13.2 months, both in the overall population (95% CI: 10.9, 16.7) and in the treatment-naïve cohort (95% CI: 7.4, 16.8).
Pharmacokinetics: The pharmacokinetics of sunitinib and sunitinib malate were evaluated in 135 healthy volunteers and 266 subjects with solid tumors.
Absorption: Maximum plasma concentrations (C
max) are generally observed between 6 - 12 hours (T
max) following oral administration. Food has no effect on the bioavailability of sunitinib.
Distribution: Binding of sunitinib and its primary active metabolite to human plasma protein
in vitro was 95% and 90%, respectively, with no apparent concentration dependence in the range of 100 - 4000 ng/mL. The apparent volume of distribution (Vd/F) for sunitinib was large (2230 L), indicating distribution into the tissues. In the dosing range of 25 - 100 mg, the area under the plasma concentration-time curve (AUC) and C
max increased proportionately with dose.
Metabolism: The calculated
in vitro Ki values for all CYP isoforms tested (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5, AND CYP4A9/11) indicated that sunitinib and its primary active metabolite are unlikely to have any clinically relevant drug-drug interactions with drugs that may be metabolized by these enzymes.
In vitro studies indicate that sunitinib neither induces nor inhibits major CYP enzymes, including CYP3A4 (see Interactions).
Sunitinib is metabolized primarily by the cytochrome P450 enzyme, CYP3A4, to produce its primary active metabolite, which is further metabolized by CYP3A4. The primary active metabolite comprises 23 to 37% of the total exposure.
Elimination: Excretion is primarily via feces (61%) with renal elimination of drug and metabolites accounting for 16% of the administered dose. Sunitinib and its primary active metabolite were the major drug-related compounds identified in plasma, urine and feces, representing 91.5%, 86.4% and 73.8% of radioactivity in pooled samples, respectively. Minor metabolites were identified in urine and feces, but generally were not found in plasma. Total oral clearance (CL/F) ranged from 34-62 L/hr with an inter-patient variability of 40%. Following administration of a single-oral dose in healthy volunteers, the terminal half-lives of sunitinib and its primary active desethyl metabolite were approximately 40-60 hours, and 80-110 hours, respectively.
Pharmacokinetics in special patient groups: Hepatic Insufficiency: Sunitinib and its primary metabolite are mainly metabolized by the liver. Systemic exposures after a single dose of sunitinib were similar in subjects with mild (Child-Pugh Class A) or moderate (Child-Pugh Class B) hepatic impairment compared to subjects with normal hepatic function. Sunitinib was not studied in subjects with severe (Child-Pugh Class C) hepatic impairment.
Renal Insufficiency: Population pharmacokinetic analyses have shown that sunitinib pharmacokinetics were unaltered in subjects with calculated creatinine clearances in the range of 42-347 mL/min. Systemic exposures after a single dose of sunitinib were similar in subjects with severe renal impairment (CL
cr<30 mL/min) compared to subjects with normal renal function (CL
cr>80 mL/min). Although sunitinib and its primary metabolite were not eliminated through hemodialysis in subjects with ESRD, the total systemic exposures were lower by 47% for sunitinib and 31% for its primary metabolite compared to subjects with normal renal function.
Cardiac Electrophysiology: QT interval prolongation was investigated in a Phase 1 trial with 24 evaluable subjects, aged 20-87 years, with advanced malignancies. At therapeutic plasma concentrations, the maximum QTcF mean change from baseline was 9.6 msec (90% CI upper limit of 15.1 msec). At approximately twice the therapeutic concentrations, the maximum QTcF mean change from baseline was 15.4 msec (90% CI upper limit of 22.4 msec). Moxifloxacin (400 mg) used as a positive control showed a 5.6 msec maximum mean QTcF change from baseline. No subjects experienced an effect on the QTc interval greater than Grade 2 (CTCAE version 3.0). No patient presented with a cardiac arrhythmia (see Precautions).
Plasma Pharmacokinetics: Following administration of a single-oral dose in healthy volunteers, the elimination half-lives of sunitinib and its primary active metabolite are approximately 40 - 60 hours, and 80 - 110 hours, respectively. With repeated daily administration, sunitinib accumulates 3- to 4-fold while the primary active metabolite accumulates 7- to 10-fold. Steady-state concentrations of sunitinib and its primary active metabolite are achieved within 10 to 14 days. By Day 14, combined plasma concentrations of sunitinib and its active metabolite are 62.9 - 101 ng/mL which are target concentrations predicted from preclinical data to inhibit receptor phosphorylation
in vitro and result in tumor stasis/growth reduction
in vivo. No significant changes in the pharmacokinetics of sunitinib or the primary, active metabolite were observed with repeated daily administration or with repeated cycles in the dosing regimens tested.
The pharmacokinetics were similar in all solid tumor populations tested and in healthy volunteers.
Population Pharmacokinetics: Population pharmacokinetic analyses of demographic data indicate that there are no clinically relevant effects of age, body weight, creatinine clearance, gender, race or ECOG score on the pharmacokinetics of sunitinib or the primary active metabolite.
Weight, performance status: Population pharmacokinetic analyses of demographic data indicate that no starting dose adjustments are necessary for weight or ECOG performance status.
Gender: Available data indicate that females could have about 30% lower apparent clearance (CL/F) of sunitinib than males; this difference, however, does not necessitate starting dose adjustments.
Toxicology: Preclinical safety data: In rat and monkey repeated-dose toxicity studies up to 9-months duration, the primary target organ effects were identified in the gastrointestinal tract (emesis and diarrhea in monkeys); adrenal gland (cortical congestion and/or hemorrhage in rats and monkeys, with necrosis followed by fibrosis in rats); hemolymphopoietic system (bone marrow hypocellularity, and lymphoid depletion of thymus, spleen, and lymph node); exocrine pancreas (acinar cell degranulation with single cell necrosis); salivary gland (acinar hypertrophy); bone joint (growth plate thickening); uterus (atrophy); and ovaries (decreased follicular development). All findings occurred at clinically relevant sunitinib plasma exposure levels. Additional effects, observed in other studies included: QTc interval prolongation, LVEF reduction, and testicular tubular atrophy, increased mesangial matrix in kidney, hemorrhage in GI tract and oral mucosa, and hypertrophy of anterior pituitary cells. Changes in the uterus (endometrial atrophy) and bone growth plate (physeal thickening or dysplasia of cartilage) are thought to be related to the pharmacological action of sunitinib. Most of these findings were reversible after 2 to 6 weeks without treatment.
Genotoxicity: The genotoxic potential of sunitinib was assessed
in vitro and
in vivo. Sunitinib was not mutagenic in bacteria using metabolic activation provided by rat liver. Sunitinib did not induce structural chromosome aberrations in human peripheral blood lymphocyte cells
in vitro. Polyploidy (numerical chromosome aberrations) was observed in human peripheral blood lymphocytes
in vitro, both in the presence and absence of metabolic activation. Sunitinib was not clastogenic in rat bone marrow
in vivo. The major active metabolite was not evaluated for genetic toxicity potential.
Carcinogenicity: In a 1-month, oral gavage dose-range finding study (0, 10, 25, 75, or 200 mg/kg/day) with continuous daily dosing in rasH2 transgenic mice, carcinoma and hyperplasia of Brunner's glands of the duodenum were observed at the highest dose (200 mg/kg/day) tested.
A 6-month, oral gavage carcinogenicity study (0, 8, 25, or 75 [reduced to 50] mg/kg/day), with daily dosing was conducted in rasH2 transgenic mice. Gastroduodenal carcinomas, an increased incidence of background hemangiosarcomas, and/or gastric mucosal hyperplasia were observed at doses of ≥25 mg/kg/day following 1- or 6-months duration (≥7.3 times the AUC in subjects administered the RDD).
In a 2-year rat carcinogenicity study (0, 0.33, 1, or 3 mg/kg/day), administration of sunitinib in 28-day cycles followed by 7-day dose-free periods resulted in increases in the incidence of pheochromocytomas and hyperplasia in the adrenal medulla of male rats given 3 mg/kg/day following >1 year of dosing (≥7.8 times the AUC in subjects administered the RDD). Brunner's glands carcinoma occurred in the duodenum at ≥1 mg/kg/day in females and at 3 mg/kg/day in males, and mucous cell hyperplasia was evident in the glandular stomach at 3 mg/kg/day in males, which occurred at ≥0.9, 7.8 and 7.8 times the AUC in subjects administered the RDD, respectively. The relevance to humans of the neoplastic findings observed in the mouse (rasH2 transgenic) and rat carcinogenicity studies with sunitinib treatment is unclear.
Reproductive and developmental toxicity: No effects on fertility were observed in male rats dosed for 58 days prior to mating with untreated females. No reproductive effects were observed in female rats treated for 14 days prior to mating with untreated males, at doses resulting in systemic exposures approximately 5 times the systemic exposure in humans. However, in repeated-dose toxicity studies performed in rats and monkeys, effects on female fertility were observed in the form of follicular atresia, degeneration of corpora lutea, endometrial changes in the uterus and decreased uterine and ovarian weights at clinically relevant systemic exposure levels. Moreover, in repeat-dose toxicity studies conducted in rats, effects on male fertility were observed in the form of tubular atrophy in the testes, reduction of spermatozoa in epididymides and colloid depletion in prostate and seminal vesicles at plasma exposure levels 25 times the systemic exposure in humans. Not all the effects observed in male rats were reversible at the end of the recovery period (6 weeks).
In rats, treatment-related embryo-fetal mortality was evident as significant reductions in the number of live fetuses, increased numbers of resorptions (early and total), corresponding increased post-implantation loss, and total litter loss in 8 of 28 pregnant females at plasma exposure levels 5.5 times the systemic exposure in humans. In rabbits, reductions in gravid uterine weights and number of live fetuses were due to increases in the number of resorptions (early and total), increases in post-implantation loss, and complete litter loss in 4 of 6 pregnant females at plasma exposure levels 3 times the systemic exposure in humans.
Sunitinib treatment in rats during organogenesis resulted in developmental effects at ≥5 mg/kg/day consisting of increased incidence of fetal skeletal malformations, predominantly characterized as retarded ossification of thoracic/lumbar vertebrae. Developmental effects in rats occurred at plasma exposure levels 6 times the systemic exposure in humans. In rabbits, developmental effects consisted of increased incidence of cleft lip at plasma exposure levels approximately equal to that observed in clinic, and cleft lip and cleft palate at plasma exposure levels 2.7 times the systemic exposure in humans.
A definitive rabbit embryo-fetal development toxicity study was not conducted as embryo-fetal effects were clearly demonstrated in the rat and reported in the preliminary study conducted in rabbits.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image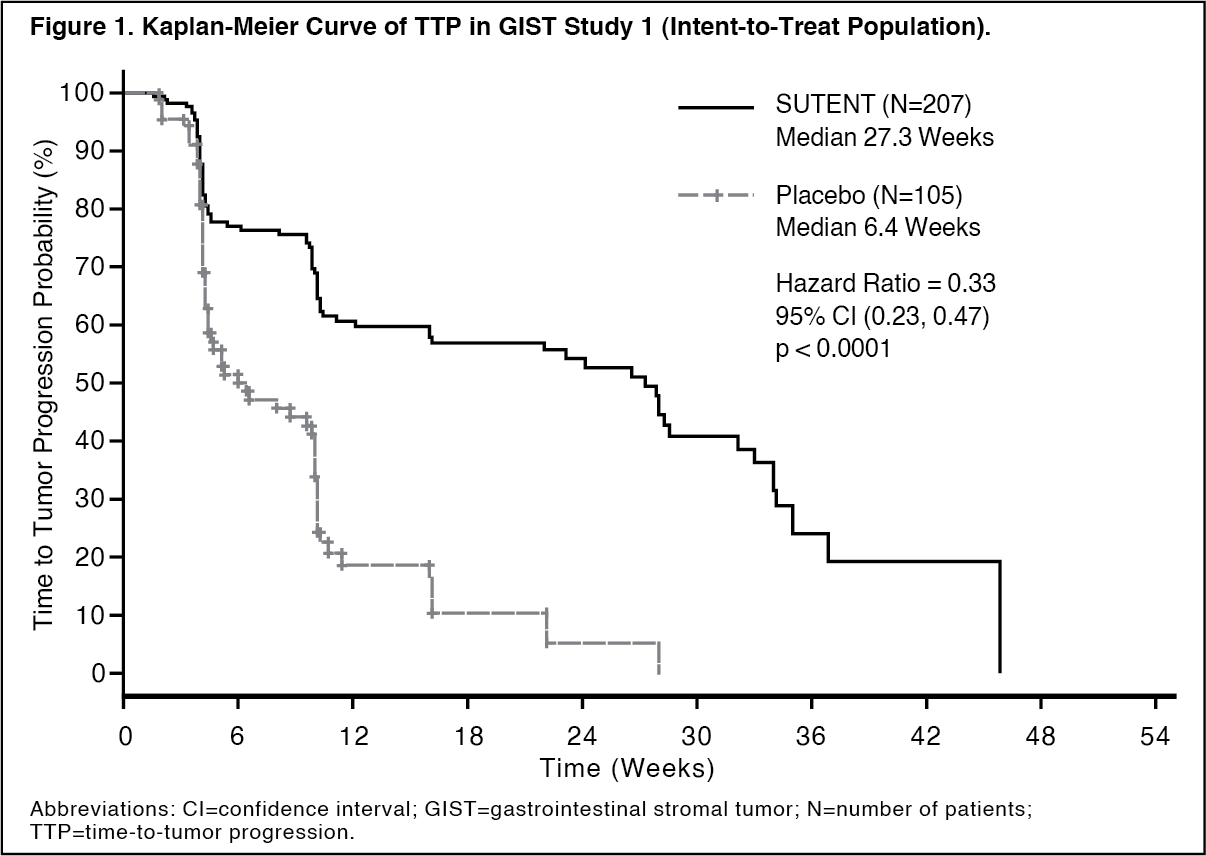 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image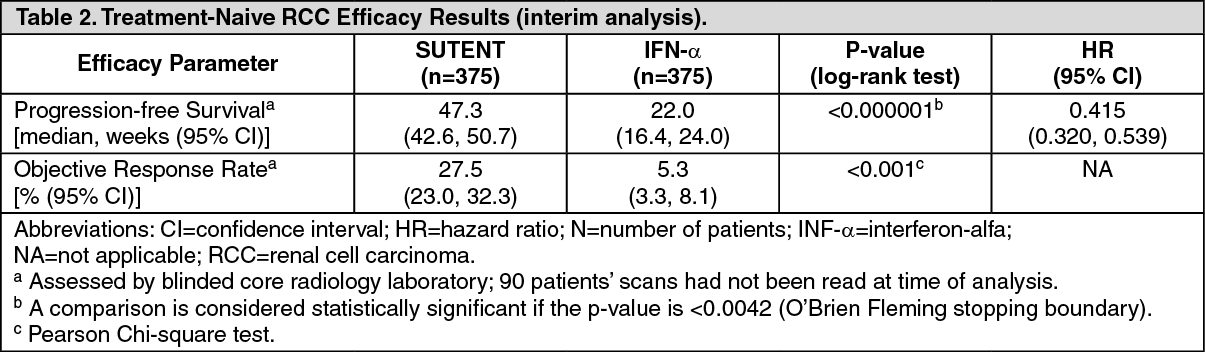 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image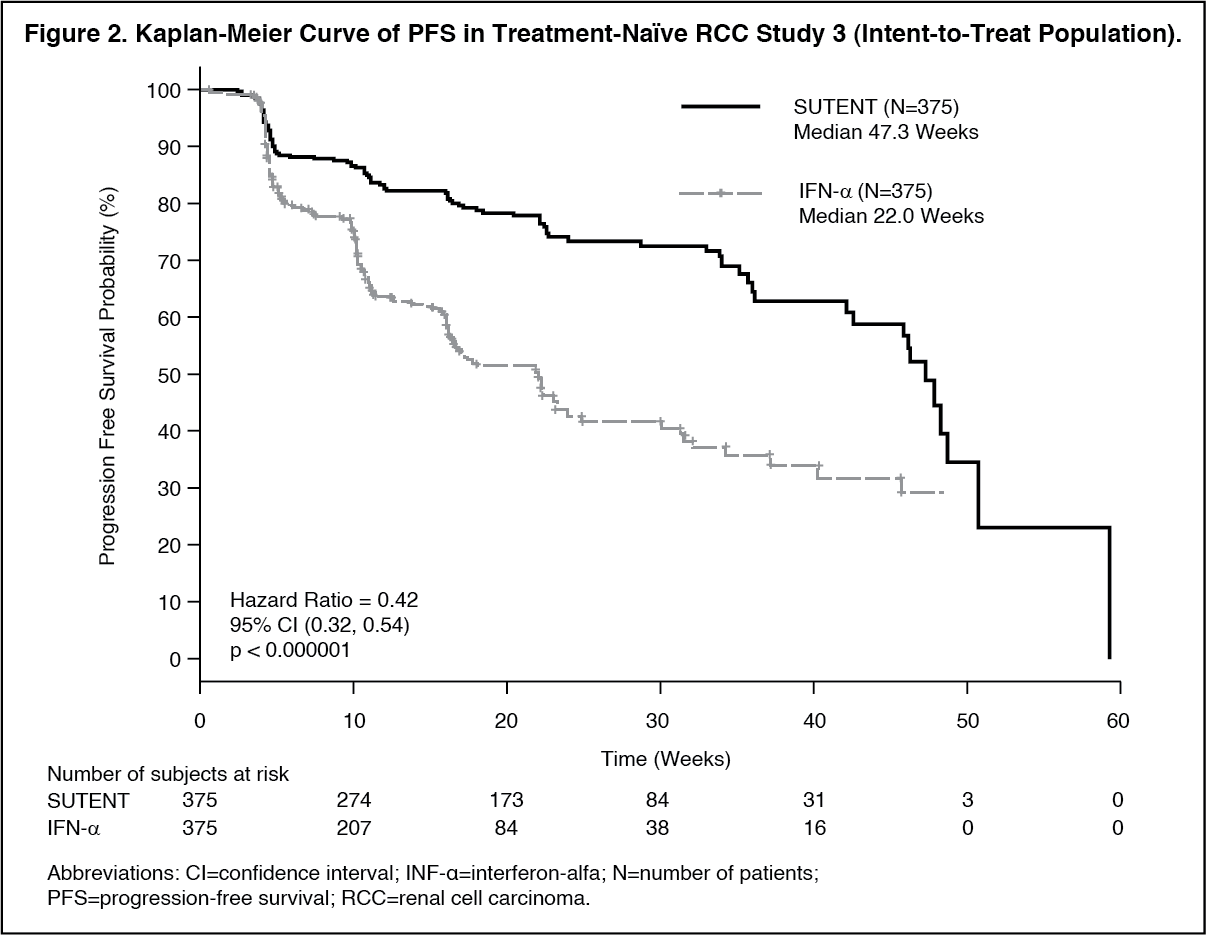 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image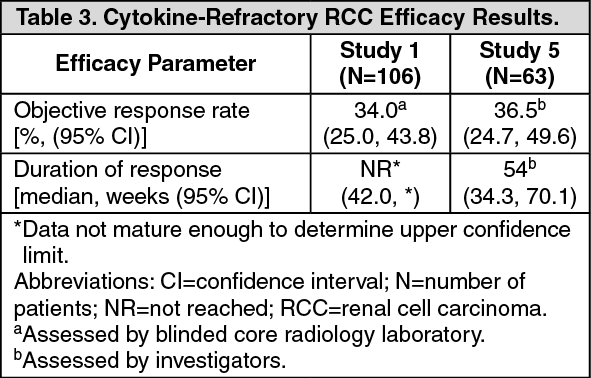 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image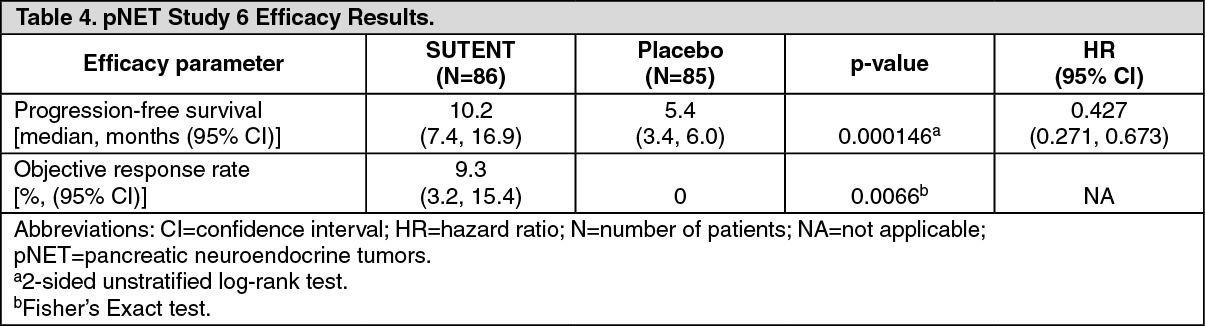 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image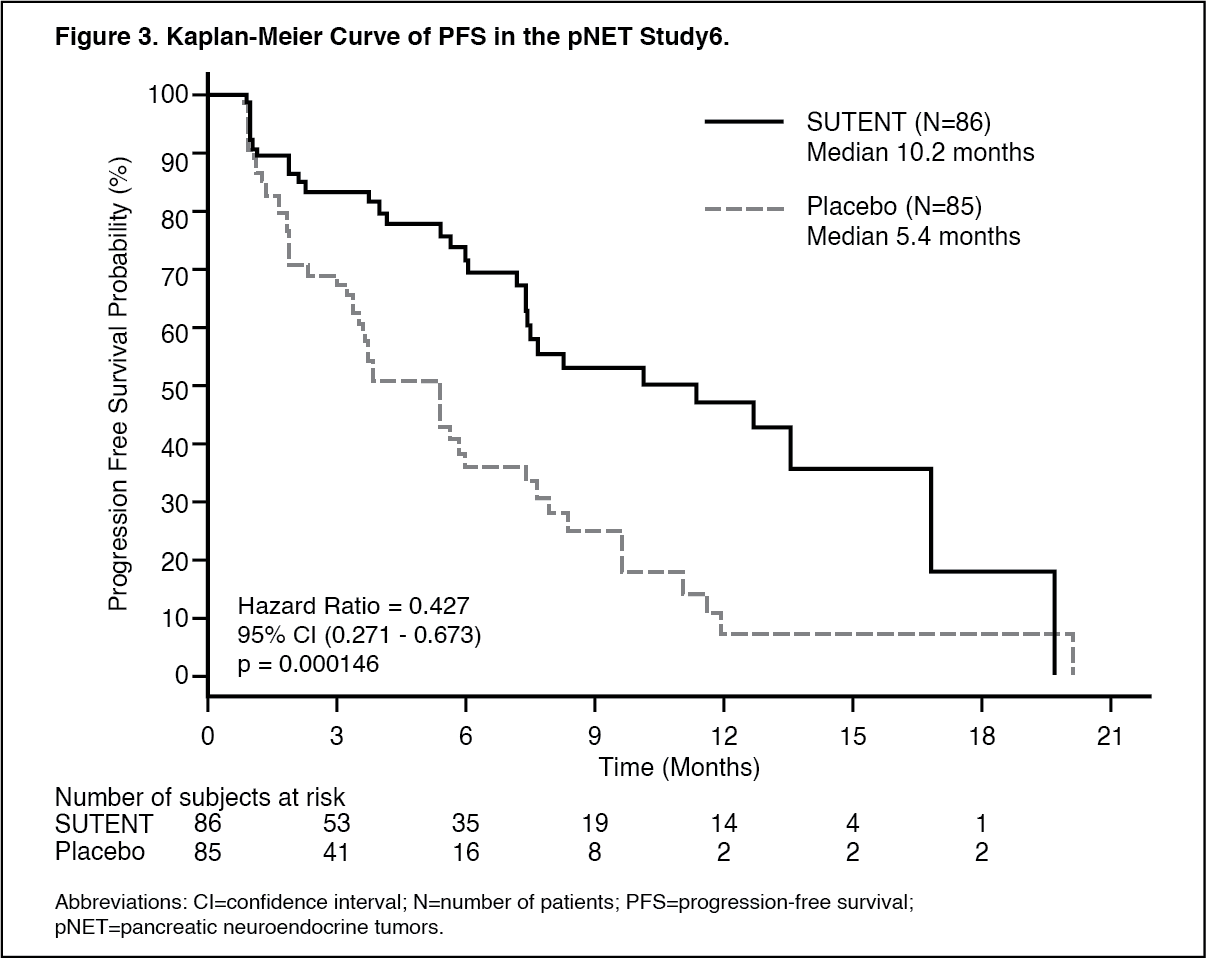 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image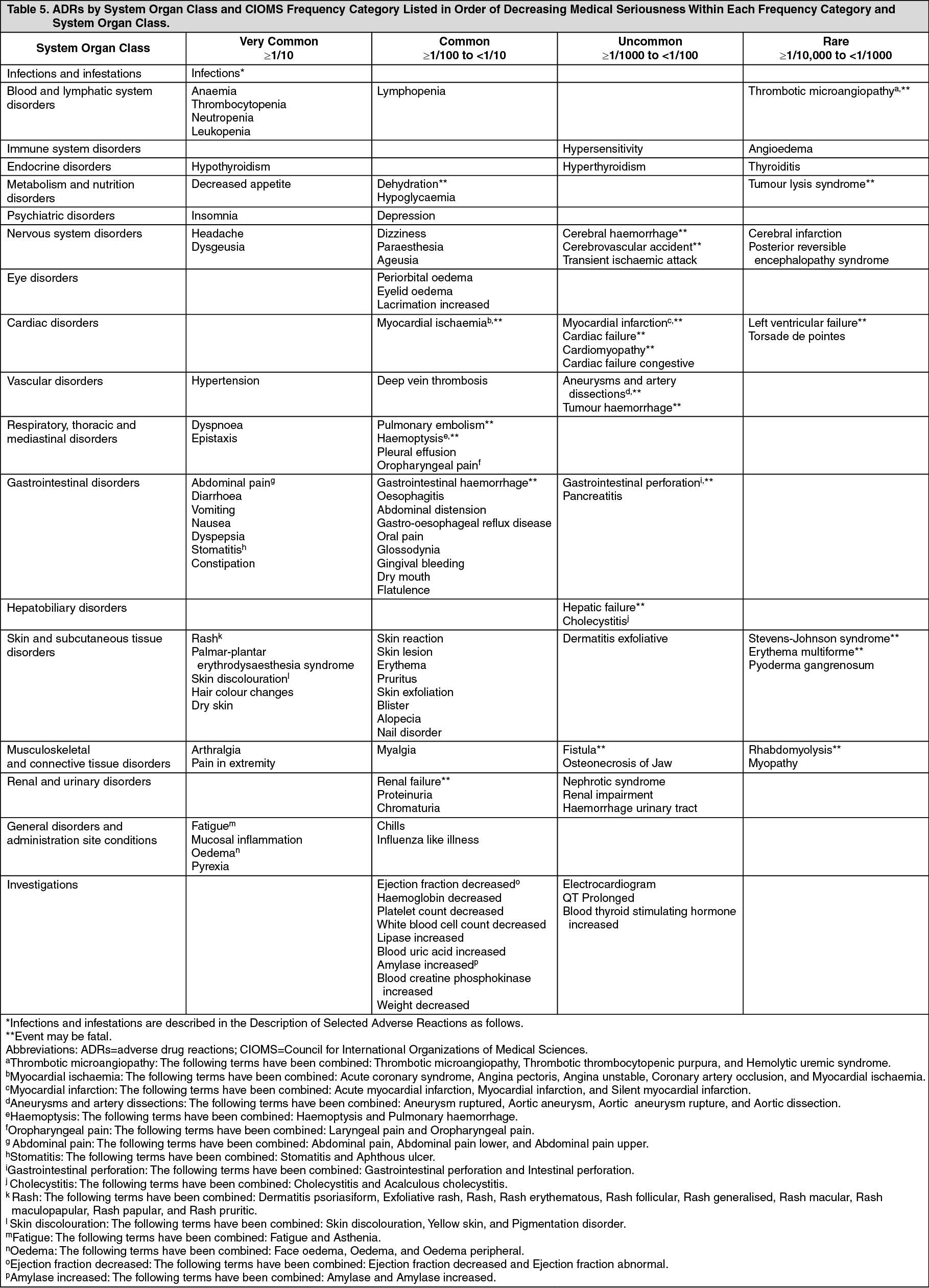 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
