


 Sign Out
Sign Out
The bactericidal action results from the interference with topoisomerase II and IV. Topoisomerases are essential enzymes which control DNA topology and assist in DNA replication, repair and transcription.
Moxifloxacin exhibits concentration dependent bactericidal killing. Minimum bactericidal concentrations are generally similar to minimum inhibitory concentrations.
Moxifloxacin is effective against β-lactam and macrolide resistant bacteria. Studies in animal models of infection have demonstrated high in vivo activity.
Resistance: Resistance mechanisms which inactivate penicillins, cephalosporins, aminoglycosides, macrolides and tetracyclines do not interfere with the antibacterial activity of moxifloxacin. There is no cross resistance between moxifloxacin and these agents. Plasmid-mediated resistance has not been observed to date.
It appears that the C8-methoxy moiety contributes to enhanced activity and lower selection of resistant mutants of gram-positive bacteria compared to the C8-H moiety. The presence of the bulky bicycloamine substituent at the C-7 position prevents active efflux, a mechanism of fluoroquinolone resistance.
In vitro studies have demonstrated that resistance to moxifloxacin develops slowly by multiple step mutations. A very low overall frequency of resistance was demonstrated (10-7 - 10-10). Serial exposure of organisms to sub-MIC concentrations of moxifloxacin showed only a small increase in MIC values.
Cross resistance among fluoroquinolones has been observed. However, some gram-positive and anaerobic organisms resistant to other fluoroquinolones are susceptible to moxifloxacin.
Effect on the intestinal flora in humans: In two volunteer studies, the following changes in the intestinal flora were seen following oral dosing with moxifloxacin. E. coli, Bacillus spp., Bacteroides vulgatus, Enterococci, and Klebsiella spp. were reduced, as were the anaerobes Bifidobacterium, Eubacterium, and Peptostreptococcus. These changes returned to normal within two weeks. Clostridium difficile toxin was not found.
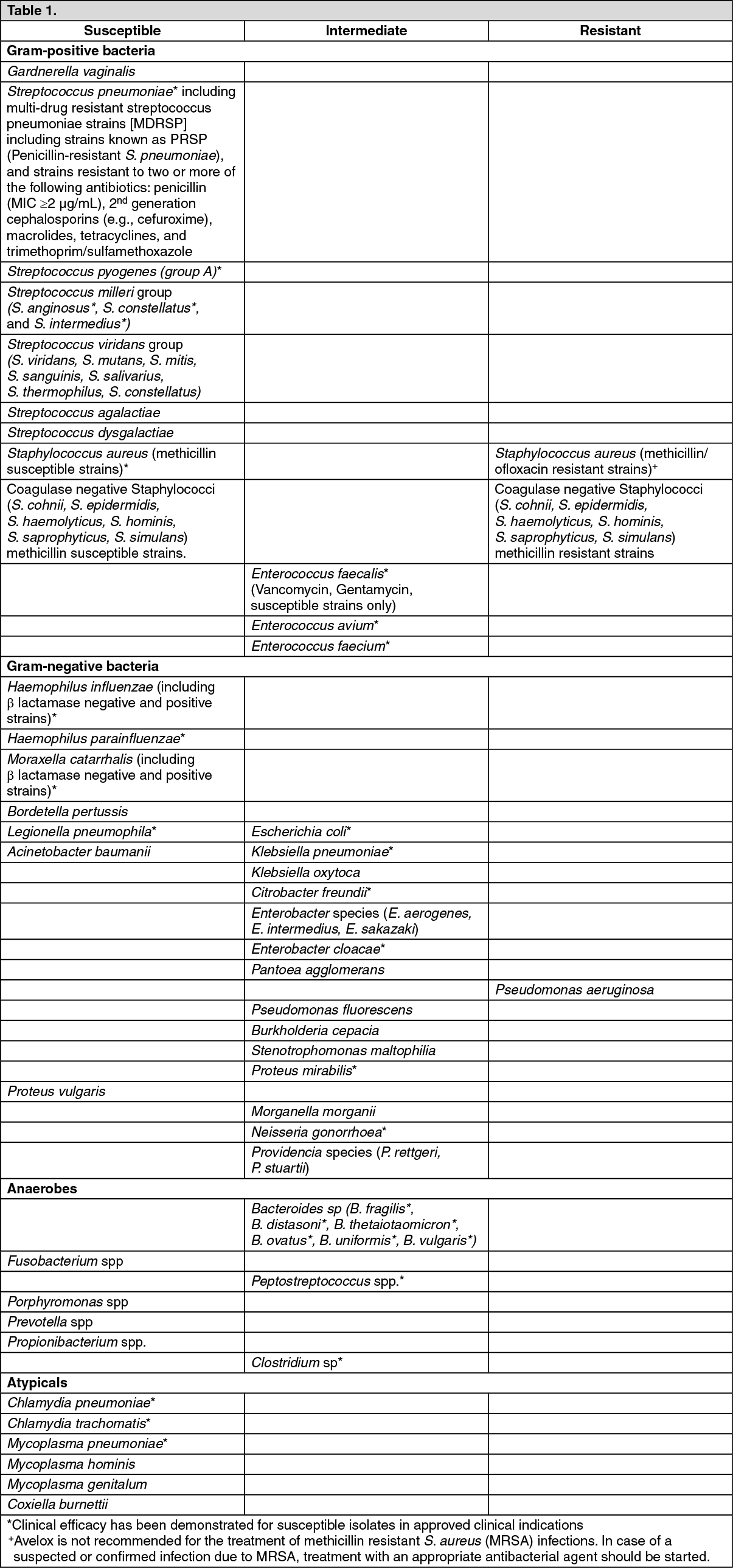
In vitro Susceptibility Data: See Table 1.
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe frequency of acquired resistance may vary geographically and with time for certain species. Local area information on resistance of organisms is desirable, particularly when treating severe infections. The previously mentioned information is provided as a guide on the probability of an organism being susceptible to moxifloxacin.
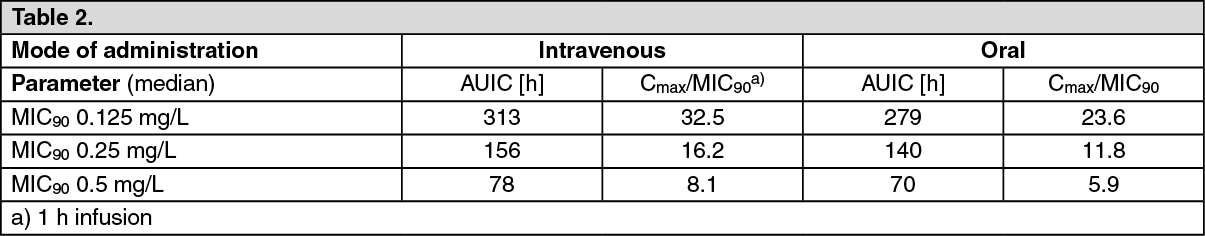
Comparison of PK/PD surrogates for intravenous and oral administration of a 400 mg Moxifloxacin (Avelox) single dose.
In patients requiring hospitalization AUC/MIC90 parameters greater than 125 and Cmax/MIC90 of 8 - 10 is predictive for clinical cure (Schentag). In outpatients these surrogate parameters are generally smaller, i.e. AUC/MIC90 greater than 30 - 40 (Dudley and Ambrose).
The following table provides the respective PK/PD surrogates for intravenous and oral administration of 400 mg moxifloxacin calculated from single dose data: See Table 2.
 Click on icon to see table/diagram/image
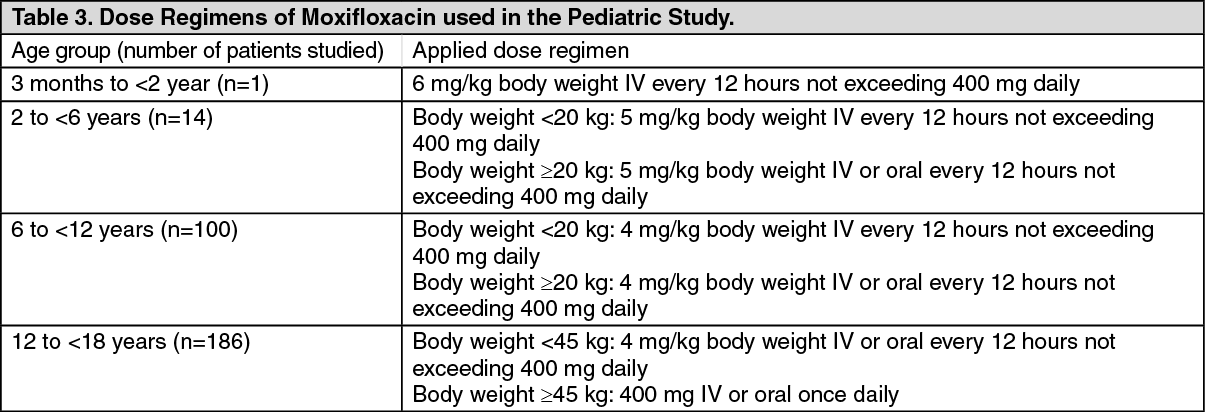
Click on icon to see table/diagram/imageSpecial patient population: Pediatric Patients: Clinical study in complicated intra-abdominal infection (cIAI): The safety of moxifloxacin in pediatric patients 3 months to <18 years of age (mean age of 12 ± 4 years) was investigated in one randomized, double-blind, active controlled trial in cIAI including appendicitis with perforation, abscesses and peritonitis. Efficacy was investigated as a secondary objective. Patients received sequential intravenous/oral moxifloxacin or intravenous ertapenem followed by oral amoxicillin/clavulanate for 5 to 14 days (mean duration was 9 days with a range of 1 to 24 days) as shown in the table as follows. (See Table 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe primary objective of the trial was to assess the safety of moxifloxacin with a focus on cardiac and musculoskeletal safety. This study enrolled 458 patients, 301 of which were treated with moxifloxacin, 150 with active control and 7 patients did not receive study treatment.
Adverse events were reported in more than half of all patients in both treatment groups. The incidence of patients experiencing at least one adverse event was similar between the treatment groups. The most frequently occurring adverse events with moxifloxacin were QT prolongation, diarrhea and phlebitis. No cardiovascular morbidity or mortality attributable to QT prolongation occurred with moxifloxacin. Discontinuation of study drug due to an adverse event was reported in 5.3% (16/301) of moxifloxacin-treated patients versus 1.3% (2/150) of comparator patients. The adverse event profile of moxifloxacin or comparator was similar across all age groups studied.
The rates of musculoskeletal events were 4.3% (13/301) in the moxifloxacin-treated group versus 3.3% (5/150) in comparator-treated patients. The majority of musculoskeletal events were reported between 12 and 53 weeks after start of study treatment, and all events resolved (clinical resolution of signs and symptoms) at the end of the study.
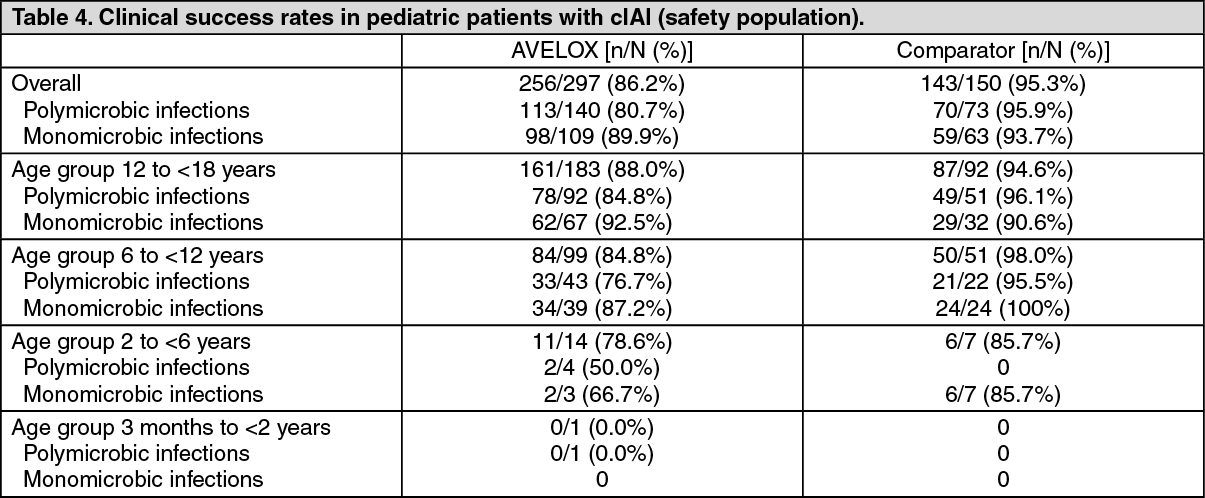
A secondary endpoint of the trial was to assess the clinical and bacteriological response at the test-of-cure visit. The clinical and bacteriological responses in children treated with moxifloxacin were within the range of that observed in adults with cIAI (see Dosage & Administration).
Descriptive analysis of the overall clinical success at the test-of-cure visit (28 to 42 days after end of treatment) in all patients treated with study drug (safety population) is shown in the table as follows. (See Table 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageSafety of moxifloxacin after single dose application: The safety and tolerability of moxifloxacin was investigated in pediatric patients 3 months to ≤14 years of age (mean age of 5 ± 4 years) in one non-randomized, non-blinded, uncontrolled, multicenter trial. The trial was designed to evaluate the pharmacokinetics of moxifloxacin. A total of 31 patients received moxifloxacin as an 1-hour intravenous infusion at different doses as shown in the table as follows. (See Table 5.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageFor results of pharmacokinetics, see Pharmacokinetics as follows. Adverse events were reported in more than half of all patients. The incidence of patients experiencing at least one adverse event (drug-related and non-drug-related) was higher in the infants and toddlers compared to children and adolescents. The most frequently occurring adverse events by MedDRA system organ class were General disorders and administration site conditions, Gastrointestinal disorders, and Skin and subcutaneous tissue disorders. There was no increase in drug-related adverse events associated with higher doses of moxifloxacin. No cardiovascular morbidity or mortality attributable to QT prolongation occurred. No musculoskeletal events were reported up to the 3-month follow-up visit.
Pharmacokinetics: Absorption and bioavailability: Following oral administration moxifloxacin is absorbed rapidly and almost completely. The absolute bioavailability amounts to approx. 91%.
Pharmacokinetics are linear in the range of 50 - 1200 mg single dose and up to 600 mg once daily dosing over 10 days. Steady state is reached within 3 days. Following a 400 mg oral dose peak concentrations of 3.1 mg/L are reached within 0.5 - 4 hours post application. Peak and trough plasma concentrations at steady state (400 mg once daily) were 3.2 and 0.6 mg/L, respectively.
Concomitant administration of moxifloxacin together with food slightly prolongs the time to reach peak concentrations by approximately 2 hours and slightly reduced peak concentrations by approximately 16%. Extent of absorption remained unchanged. As AUC/MIC is most predictive for antimicrobial efficacy of fluoroquinolones, this effect is clinically not relevant. Therefore, Moxifloxacin (Avelox) can be administered independently from meals.
After a single 400 mg intravenous 1 hour infusion peak concentrations of approximately 4.1 mg/L were reached in the plasma at the end of infusion which corresponds to a mean increase of approx. 26 % relative to the oral application. Exposure to drug in terms of AUC at a value of approximately 39 mg*h/L is only slightly higher compared to the exposure after oral administration (35 mg*h/L) in accordance with the absolute bioavailability of approximately 91%.
Following multiple intravenous dosing (1hour infusion), peak and trough plasma concentrations at steady state (400 mg once daily) were between 4.1 to 5.9 and 0.43 to 0.84 mg/L respectively. At steady-state the exposure to drug within the dosing interval is approximately 30 % higher than after the first dose. In patients mean steady state concentrations of 4.4 mg/L were observed at the end of a 1-hour infusion.
Distribution: Moxifloxacin is distributed very rapidly to extra vascular spaces. Exposure to drug in terms of AUC (AUCnorm = 6 kg*h/L) is high with a volume of distribution at steady state (Vss) of approximately 2 L/kg. In saliva peak concentrations higher than those of plasma may be reached. In in vitro and ex vivo experiments over a range of 0.02 to 2 mg/L a protein binding of approximately 45 % independent from the concentration of the drug was determined. Moxifloxacin is mainly bound to serum albumin. Due to this low value high free peak concentrations > 10x MIC are observed.
Moxifloxacin reaches high concentrations in tissues like lung (epithelial fluid, alveolar macrophages, biotic tissue), the sinuses (maxillary and ethmoid sinus, nasal polypi) and inflamed lesions (cantharide blister fluid) where total concentrations exceeding those of the plasma concentrations are reached. High free drug concentrations are measured in interstitial body water (saliva, intramuscular, subcutaneous). In addition, high drug concentrations were detected in abdominal tissues and fluids and female genital tract.
The peak concentrations and site vs. plasma concentration ratios for various target tissues yielded comparable results for both modes of drug administration after a single dose of 400 mg moxifloxacin.
Metabolism: Moxifloxacin undergoes Phase II biotransformation and is excreted via renal and biliary/faecal pathways as unchanged drug as well as in form of a sulfo-compound (M1) and a glucuronide (M2). M1 and M2 are the only metabolites relevant in humans, both are microbiologically inactive. Neither in in vitro nor in clinical Phase I studies metabolic pharmacokinetic interactions with other drugs undergoing Phase I biotransformation involving Cytochrome P-450 enzymes were observed.
Independent from the route of administration the metabolites M1 and M2 are found in the plasma at concentrations lower than the parent drug. Preclinical investigations adequately covered both metabolites thus excluding potential implications with respect to safety and tolerability.
Elimination: Moxifloxacin is eliminated from plasma with a mean terminal half life of approximately 12 hours. The mean apparent total body clearance following a 400 mg dose ranges from 179 to 246 mL/min. Renal clearance amounted to about 24 - 53 mL/min suggesting partial tubular reabsorption of the drug from the kidneys. Concomitant administration of ranitidine and probenecid did not alter renal clearance of the drug.
Mass balance of the mother compound and Phase II metabolites of moxifloxacin yielded an almost complete recovery of approximately 96-98% independent from the route of administration with no indication of oxidative metabolism.
Geriatric patients: Pharmacokinetics of moxifloxacin are not affected by age.
Gender: There was a 33% difference in the pharmacokinetics (AUC, Cmax) of moxifloxacin between male and female subjects. Drug absorption was unaffected by gender. These differences in the AUC and Cmax were attributable to the differences in body weight rather than gender. They are not considered as clinically relevant.
Ethnic differences: Possible interethnic differences were examined in Caucasian, Japanese, Black and other ethnic groups. No clinically relevant interethnic differences in pharmacokinetics could be detected.
Pediatric Patients: Pharmacokinetics of moxifloxacin were studied after single dose intravenous (i.v.) administration (1 h infusion) in pediatric patients ranging from 3 months to <14 years. The corresponding PK parameters are summarized in the subsequent table. (See Table 6.)
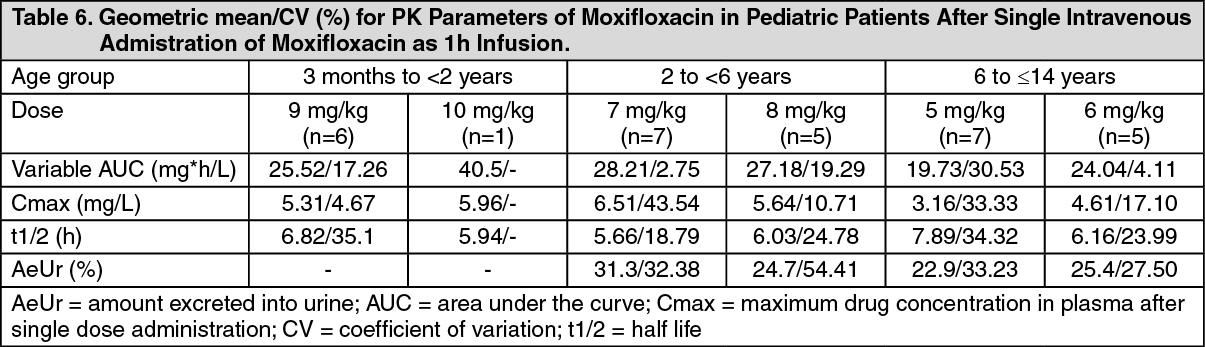 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThese data were used to derive oral and i.v. dosing schemes for a clinical Phase III study in pediatric patients as previously mentioned in Pharmacodynamics. A population PK analysis of data from Phase III with i.v. and sequential i.v. to oral treatment confirmed that the dosing schemes lead to a systemic moxifloxacin exposure which is comparable to the exposure seen in adult subjects. This is in the effective range for antimicrobial treatment based on PK/PD considerations (AUC/MIC). For pediatric patients <12 years of age and <45 kg body weight a switch from once daily to a bid dosing is suitable to avoid overly high peak concentrations. The absolute bioavailability of moxifloxacin was estimated at 86%.
Patients with renal impairment: The pharmacokinetics of moxifloxacin are not significantly changed by renal impairment (including creatinine clearance < 30 mL/min/1.73 m2) and in patients on chronic dialysis i.e. hemodialysis and continuous ambulatory peritoneal dialysis.
Patients with hepatic impairment: Moxifloxacin plasma concentrations of patients with mild to severe hepatic impairment (Child Pugh A to C) did not reveal clinically relevant differences compared to healthy volunteers or patients with normal hepatic function, respectively (see "Precautions" in patients with liver cirrhosis).
Toxicology: Preclinical safety data: In a local tolerability study performed in dogs, no signs of local intolerability were seen when moxifloxacin was administered intravenously. After intra-arterial injection inflammatory changes involving the peri-arterial soft tissue were observed suggesting that intra-arterial administration of moxifloxacin should be avoided.
Carcinogenicity, Mutagenicity: Although conventional long-term studies to determine the carcinogenic potential of moxifloxacin have not been performed, the drug has been subject to a range of in vitro and in vivo genotoxicity tests. In addition, an accelerated bioassay for human carcinogenesis (initiation/promotion assay) was performed in rats. Negative results were obtained in 4 strains of the Ames test, in the HPRT mutation assay in Chinese hamster ovary cells and in the UDS assay in rat primary hepatocytes. As with other fluoroquinolones the Ames test with TA 102 was positive and the in vitro test in the Chinese hamster v79 cells showed chromosomal abnormalities at high concentrations (300 mcg/mL). However, the in vivo micronucleus assay in the mouse was negative. An additional in vivo assay, the dominant lethal assay in the mouse, was negative as well. It is concluded that the negative in vivo results adequately reflect the in vivo situation in terms of genotoxicity. No evidence of carcinogenicity was found in an initiation/promotion assay in rats.
ECG: At high concentrations, moxifloxacin is an inhibitor of the delayed rectifier potassium current of the heart and may thus cause prolongations of the QT-interval. Toxicological studies performed in dogs using oral doses of ≥ 90 mg/kg leading to plasma concentrations ≥ 16 mg/L caused QT-prolongations, but no arrhythmias. Only after very high cumulative intravenous administration of more than 50 fold the human dose (> 300 mg/kg), leading to plasma concentrations of ≥ 200 mg/L (more than 30 fold the therapeutic level after intravenous administration), reversible, non-fatal ventricular arrhythmias were seen.
Arthrotoxicity: Fluoroquinolones are known to cause lesions in the cartilage of the major diarthrodial joints in immature animals. The lowest oral dose of moxifloxacin causing joint toxicity in juvenile dogs was four times maximum recommended therapeutic dose (400 mg/50 kg person) on a mg/kg basis, with plasma concentrations two to three times higher than those at the recommended therapeutic dose.
Reprotoxicity: Reproductive studies performed in rats, rabbits and monkeys indicate that placental transfer of moxifloxacin occurs. Studies in rats (per os and i.v.) and monkeys (per os) did not show evidence of teratogenicity or impairment of fertility following administration of moxifloxacin. Skeletal malformations were observed in rabbits that had been treated with an intravenous dose of 20 mg/kg. This study result is consistent with the known effects of fluoroquinolones on skeletal development (see "Use in Pregnancy & Lactation"). There was an increase in the incidence of abortions in monkeys and rabbits at human therapeutic concentrations. In rats, decreased foetal weights, an increased prenatal loss, a slightly increased duration of pregnancy and an increased spontaneous activity of some male and female offspring was observed at doses which were 63 times the maximum recommended dose on a mg/kg basis with plasma concentrations in the range of the human therapeutic dose.