


 Sign Out
Sign Out


Non-melanoma skin cancer: Based on an integrated analysis of the randomized, controlled phase 3 studies (PCYC-1112-CA, PCYC-1115-CA, CLL3001, PCYC-1130-CA, MCL3001, PCYC-1127-CA, E1912, and CLL3011), the incidence of non-melanoma skin cancer was 5% in IMBRUVICA-treated patients and 2% in comparator-treated patients.
Mantle cell lymphoma: The data described as follows reflect exposure to Ibrutinib (Imbruvica) in a phase 2 clinical study (PCYC‑1104‑CA) and a randomized phase 3 study (MCL3001) in patients with MCL (n=250).
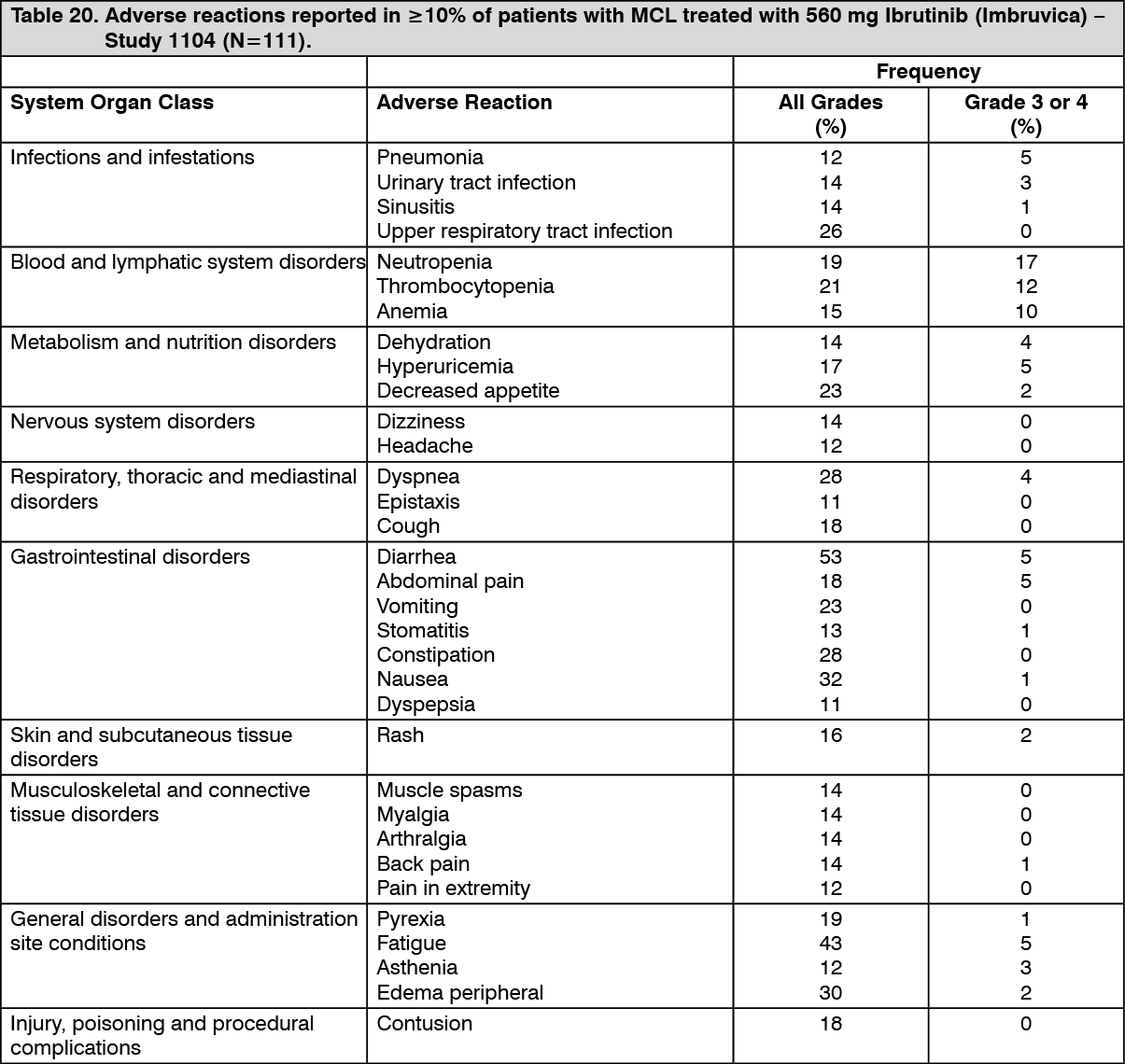
The most commonly occurring adverse reactions for MCL (≥20%) were diarrhea, hemorrhage (e.g., bruising), fatigue, musculoskeletal pain, nausea, upper respiratory tract infection, cough, and rash.
The most common Grade 3/4 adverse reactions (≥5%) were: neutropenia, thrombocytopenia, pneumonia, and anemia.
Discontinuation and dose reduction due to ARs: Of the 250 patients treated with Ibrutinib (Imbruvica) for MCL, seven (3%) discontinued treatment due to adverse reactions. The most frequent adverse reactions leading to treatment discontinuation included hemorrhage, pneumonia, and thrombocytopenia. Adverse reactions leading to dose reduction occurred in 6% of patients.
Adverse reactions from Study 1104 are described as follows in Table 20 to reflect exposure to Ibrutinib (Imbruvica) in patients with MCL who received at least one prior therapy with a median treatment duration of 8.3 months. (See Table 20.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageSerious adverse reactions: In the phase 2 study, serious adverse reactions were reported in 60% of patients (treatment-emergent frequencies). Serious adverse reactions that occurred in greater than 2% of patients were atrial fibrillation (6%), pneumonia (5%), urinary tract infection (4%), abdominal pain (3%), subdural hematoma (3%), febrile neutropenia (3%), acute renal failure (3%), peripheral edema (3%), and pyrexia (3%).
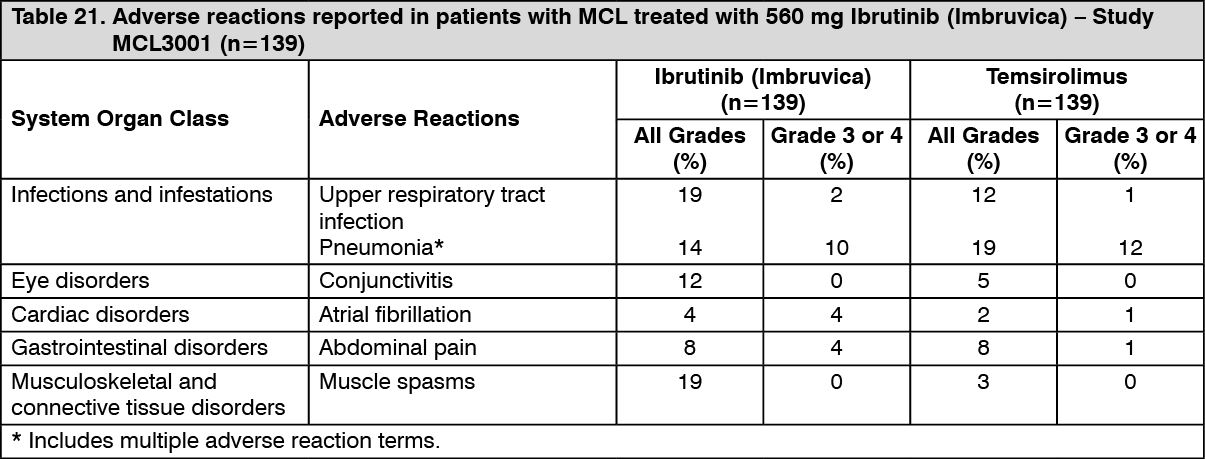
Adverse reactions from Study MCL3001 are described as follows in Table 21 reflecting exposure to Ibrutinib (Imbruvica) in patients with MCL who received at least one prior therapy, treated with a median treatment duration of 14.4 months. (See Table 21.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageChronic lymphocytic leukemia/Small lymphocytic lymphoma: The data described as follows reflect exposure to Ibrutinib (Imbruvica) in two single arm, open-label clinical studies (Study PCYC-1102-CA and PCYC-1142-CA) and six randomized clinical studies (Study PCYC-1115-CA, Study PCYC-1112-CA, Study CLL3001, PCYC-1130-CA, E1912, and CLL3011) in patients with CLL/SLL (n=1562).
The most commonly occurring adverse reactions in studies PCYC-1102-CA, PCYC-1142-CA, PCYC-1112-CA, PCYC-1115-CA, CLL3001, and PCYC-1130-CA, E1912, and CLL3011 (≥ 20%) were diarrhea, neutropenia, musculoskeletal pain, rash, thrombocytopenia, hemorrhage (e.g., bruising), nausea, arthralgia, headache, upper respiratory tract infection, and pyrexia.
The most common Grade 3/4 adverse reactions (≥5%) were: neutropenia, lymphocytosis, thrombocytopenia, hypertension, and pneumonia.
Discontinuation and dose reduction due to ARs: Six percent of patients receiving Ibrutinib (Imbruvica) in studies PCYC-1102-CA, PCYC-1142-CA, PCYC-1112-CA, PCYC-1115-CA, CLL3001, PCYC-1130-CA, E1912, and CLL3011 discontinued treatment due to adverse reactions. The most frequent adverse reactions leading to treatment discontinuation included pneumonia, atrial fibrillation, neutropenia, and rash. Adverse reactions leading to dose reduction occurred in approximately 9% of patients.
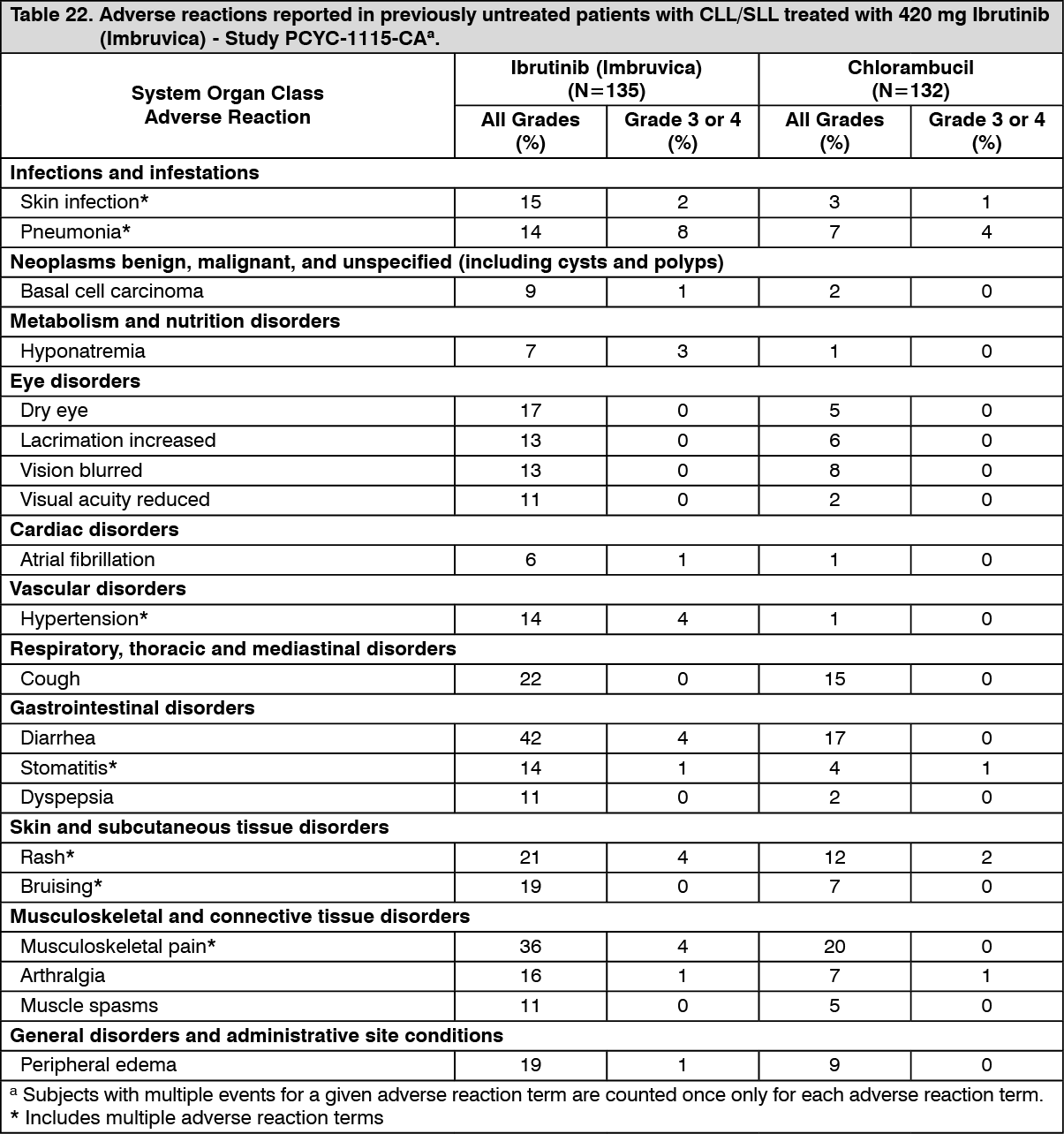
Patients with previously untreated CLL/SLL: Single agent: Adverse reactions described as follows in Table 22 reflect exposure to Ibrutinib (Imbruvica) with a median duration of 17.4 months, which is approximately 2.5 times the median exposure to chlorambucil of 7.1 months in Study PCYC-1115-CA. (See Table 22.)
 Click on icon to see table/diagram/image
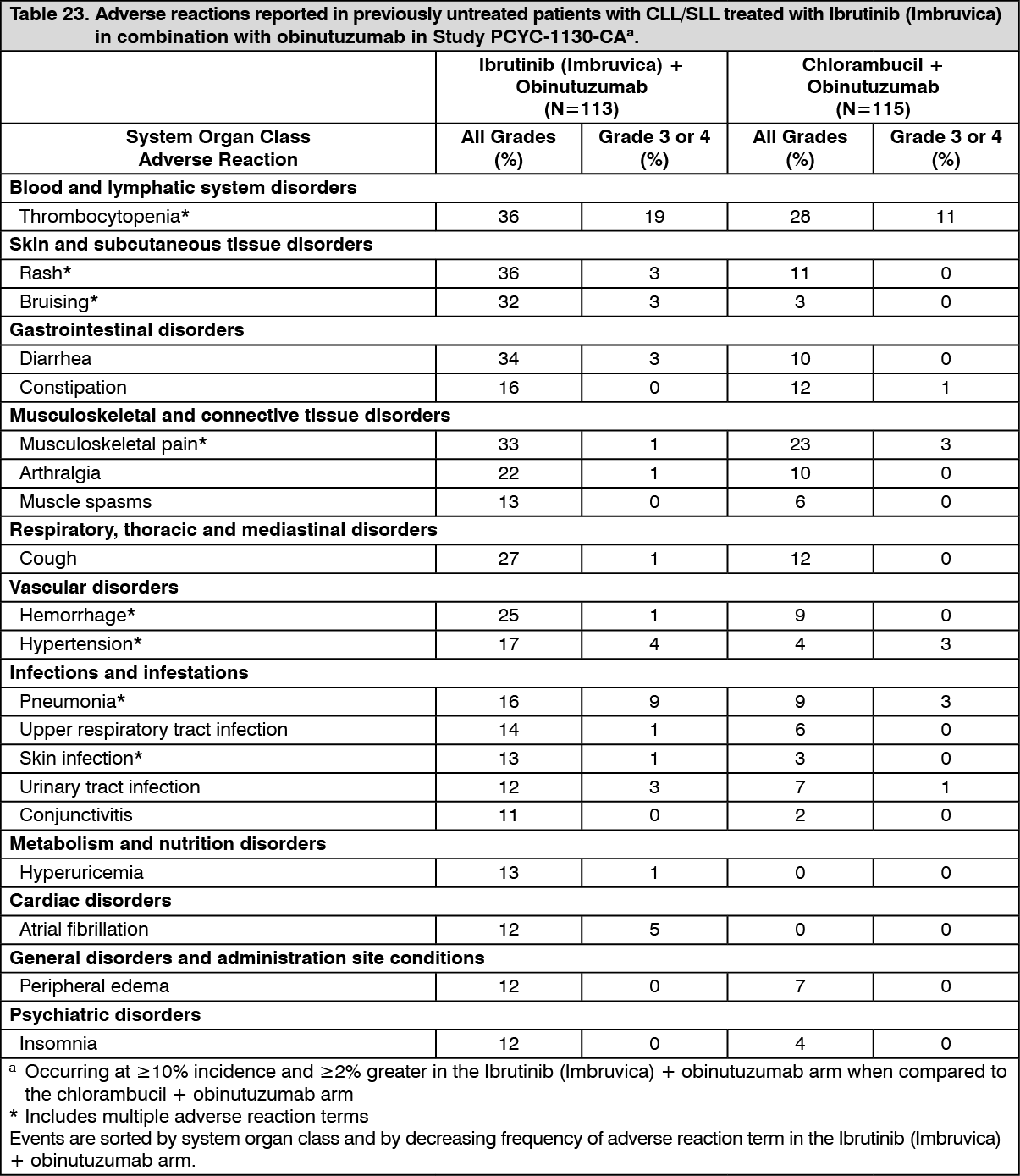
Click on icon to see table/diagram/imageCombination therapy: Adverse reactions described as follows in Table 23 reflect exposure to Ibrutinib (Imbruvica) + obinutuzumab with a median duration of 29.3 months and exposure to chlorambucil + obinutuzumab with a median duration of 5.1 months in Study PCYC-1130-CA. (See Table 23.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAdverse reactions and laboratory abnormalities described as follows in Tables 24 and 25 reflect exposure to Ibrutinib (Imbruvica) in combination with rituximab (IR) or received fludarabine, cyclophosphamide, and rituximab (FCR) with a median duration of 34.3 months for IR and 4.7 months for FCR in Study E1912. (See Tables 24 and 25.)
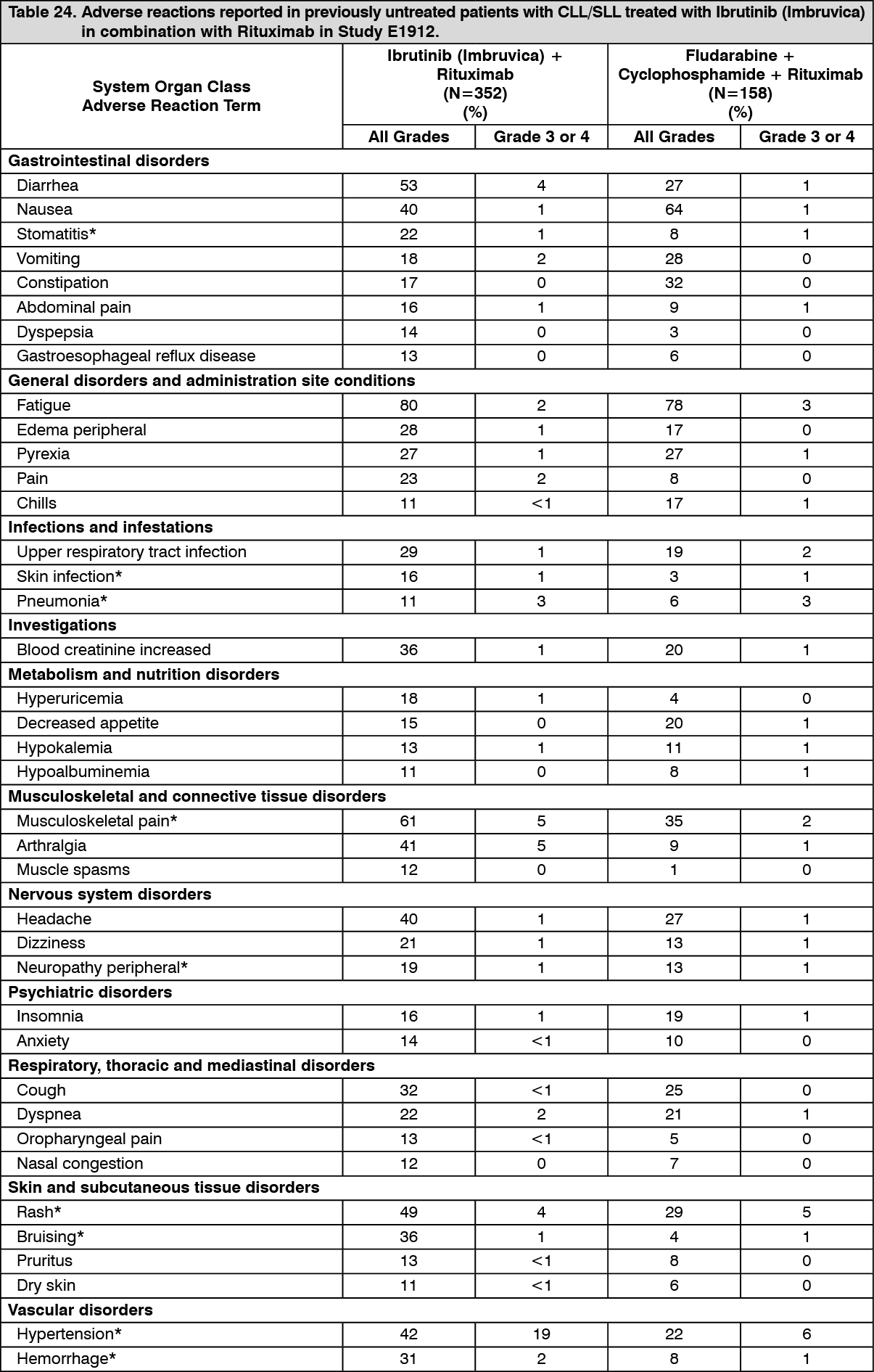 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image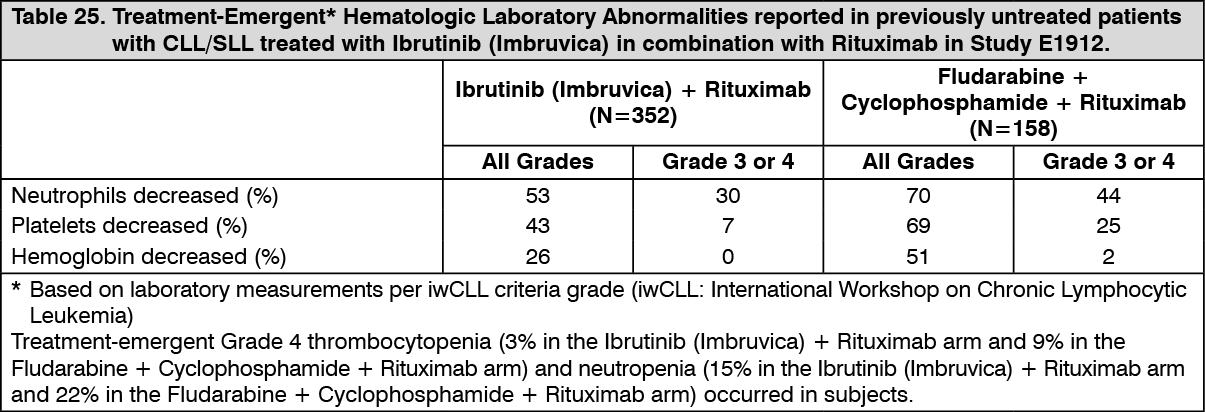 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAdverse reactions and laboratory abnormalities described as follows in Tables 26 and 27 reflect exposure to Ibrutinib (Imbruvica) in combination with venetoclax with a median duration of 14.1 months in patients with previously untreated CLL/SLL who were 70 years or younger in Study PCYC-1142-CA. (See Tables 26 and 27.)
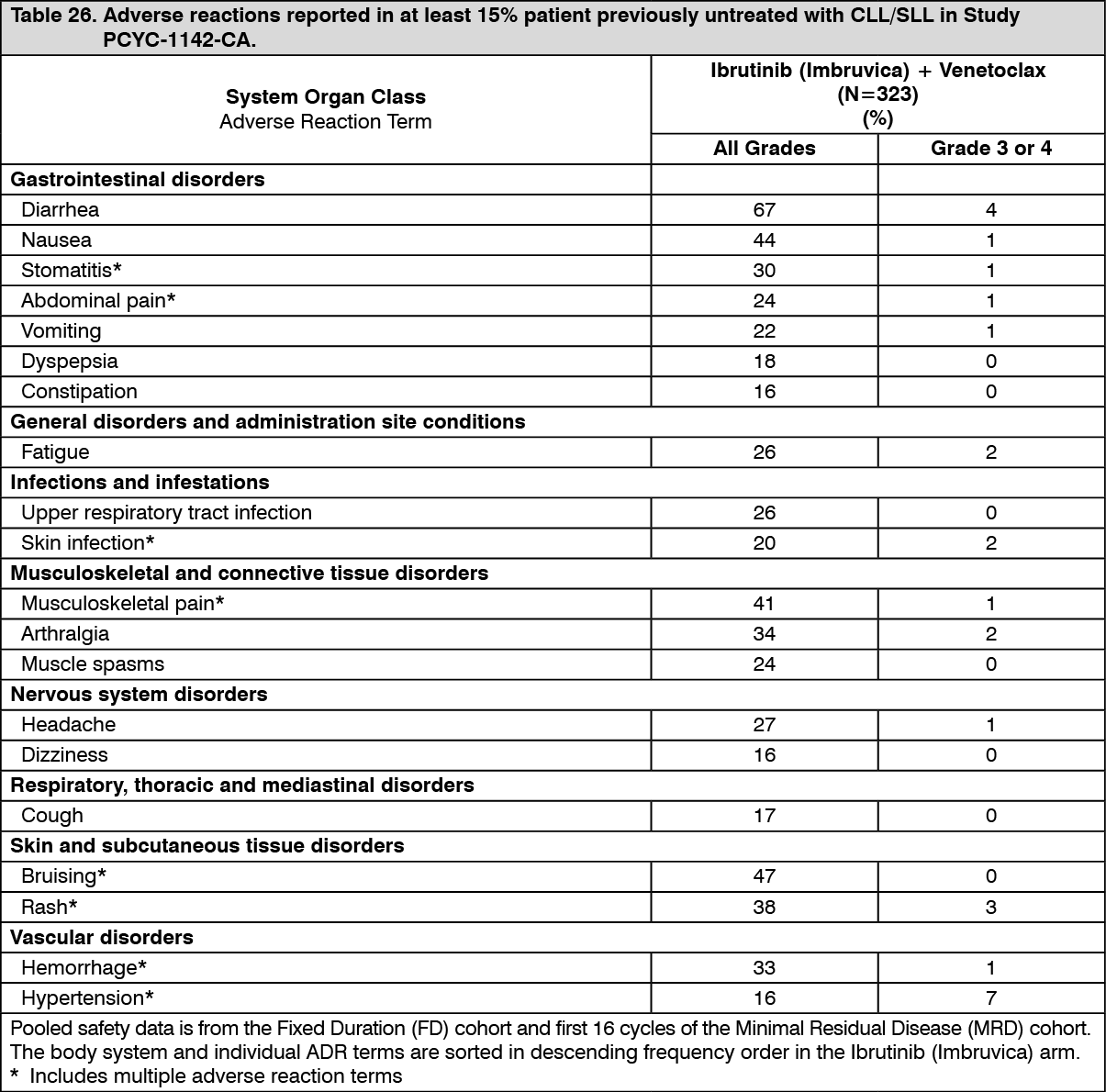 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image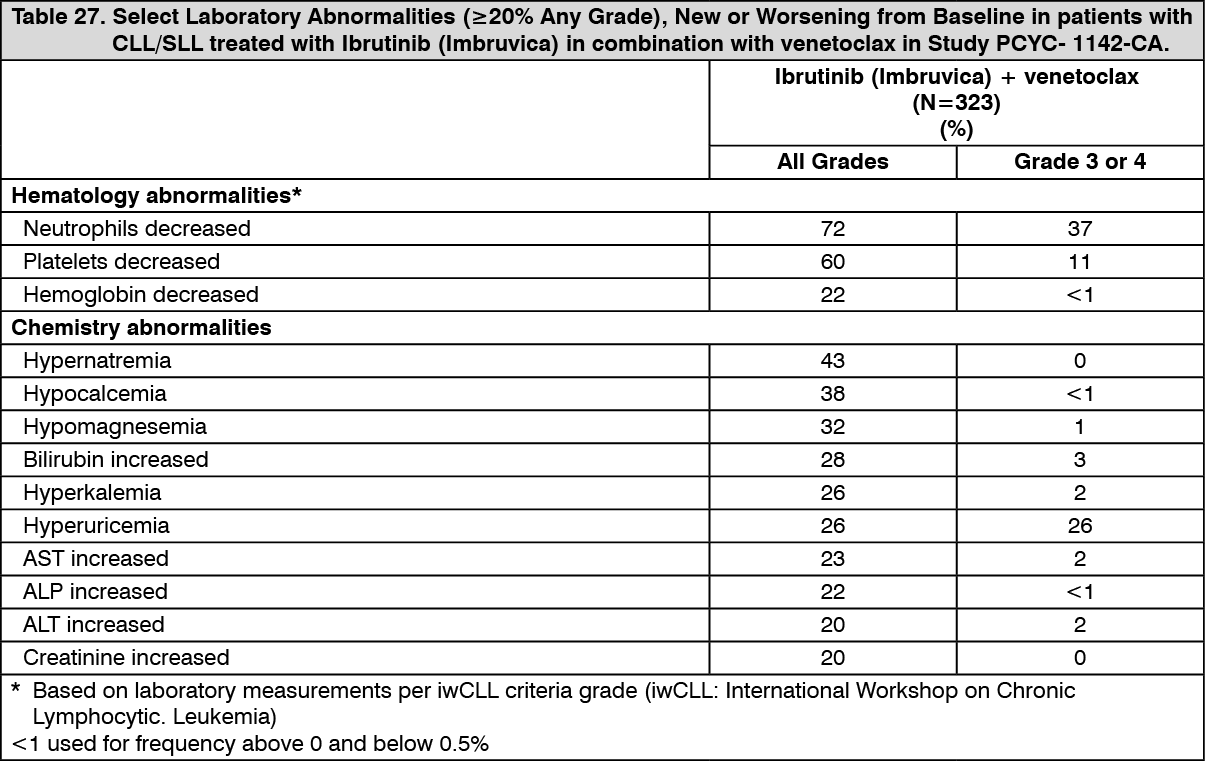 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAdverse reactions and laboratory abnormalities described as follows in Tables 28 and 29 reflect exposure to Ibrutinib (Imbruvica) + venetoclax with a median duration of 13.8 months and exposure to chlorambucil + obinutuzumab with a median of 5.1 months in Study CLL3011 in patients with previously untreated CLL/SLL who were 65 years or older, or adult patients <65 years of age with a CIRS score >6 or CrCL <70 mL/min. (See Tables 28 and 29.)
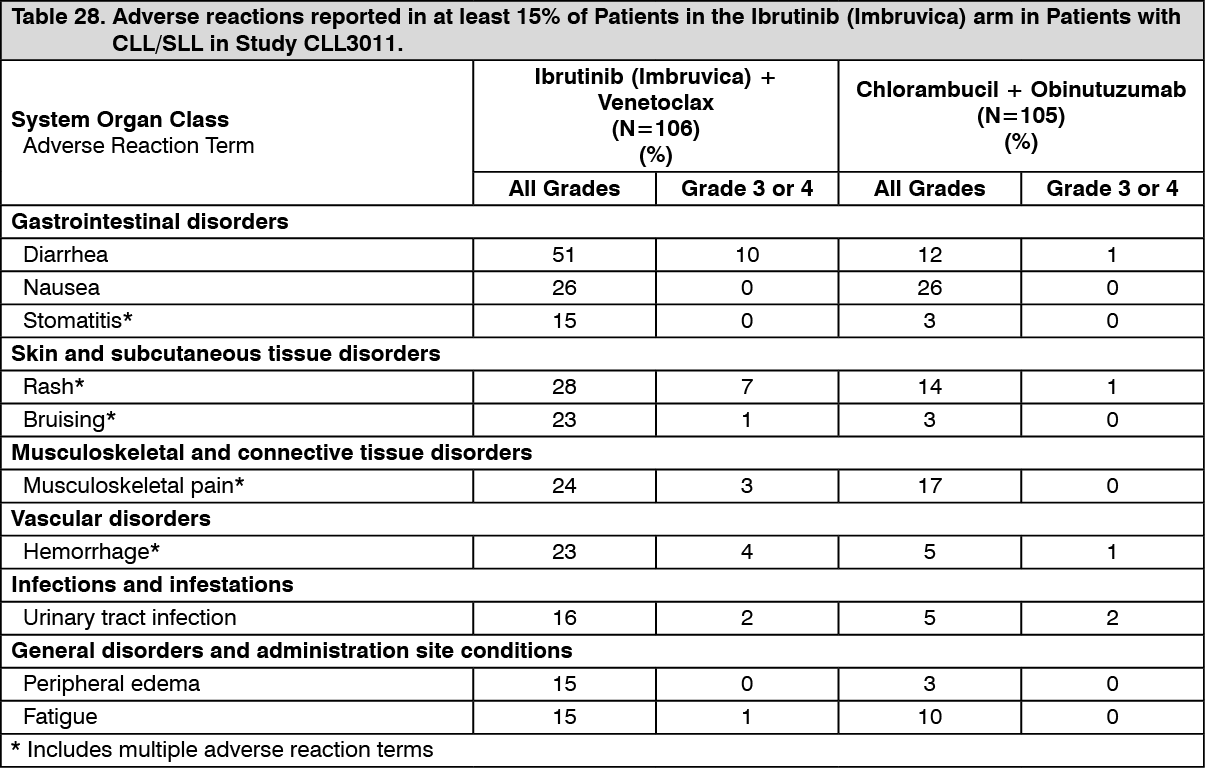 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image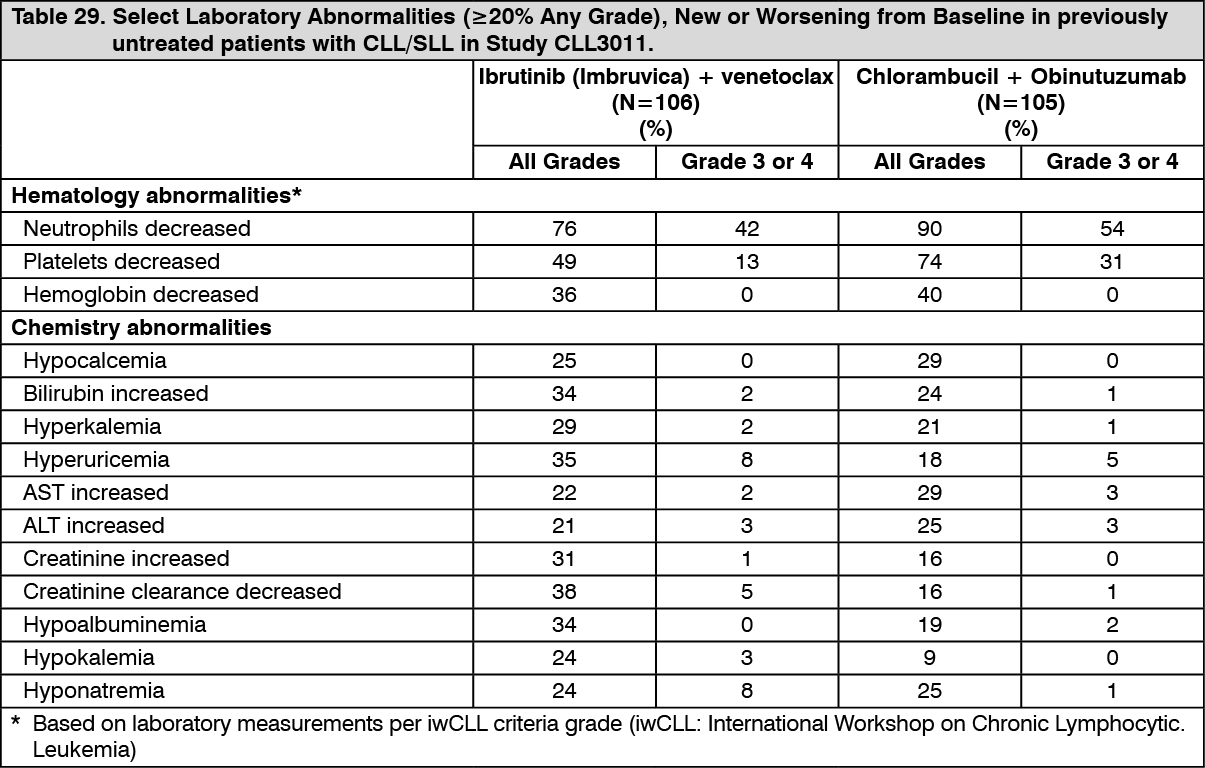 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePatients with CLL/SLL who received at least one prior therapy: Single agent: Adverse reactions described in Table 30 as follows reflect exposure to Ibrutinib (Imbruvica) with a median duration of 8.6 months and exposure to ofatumumab with a median duration of 5.3 months in Study PCYC-1112-CA. (See Table 30.)
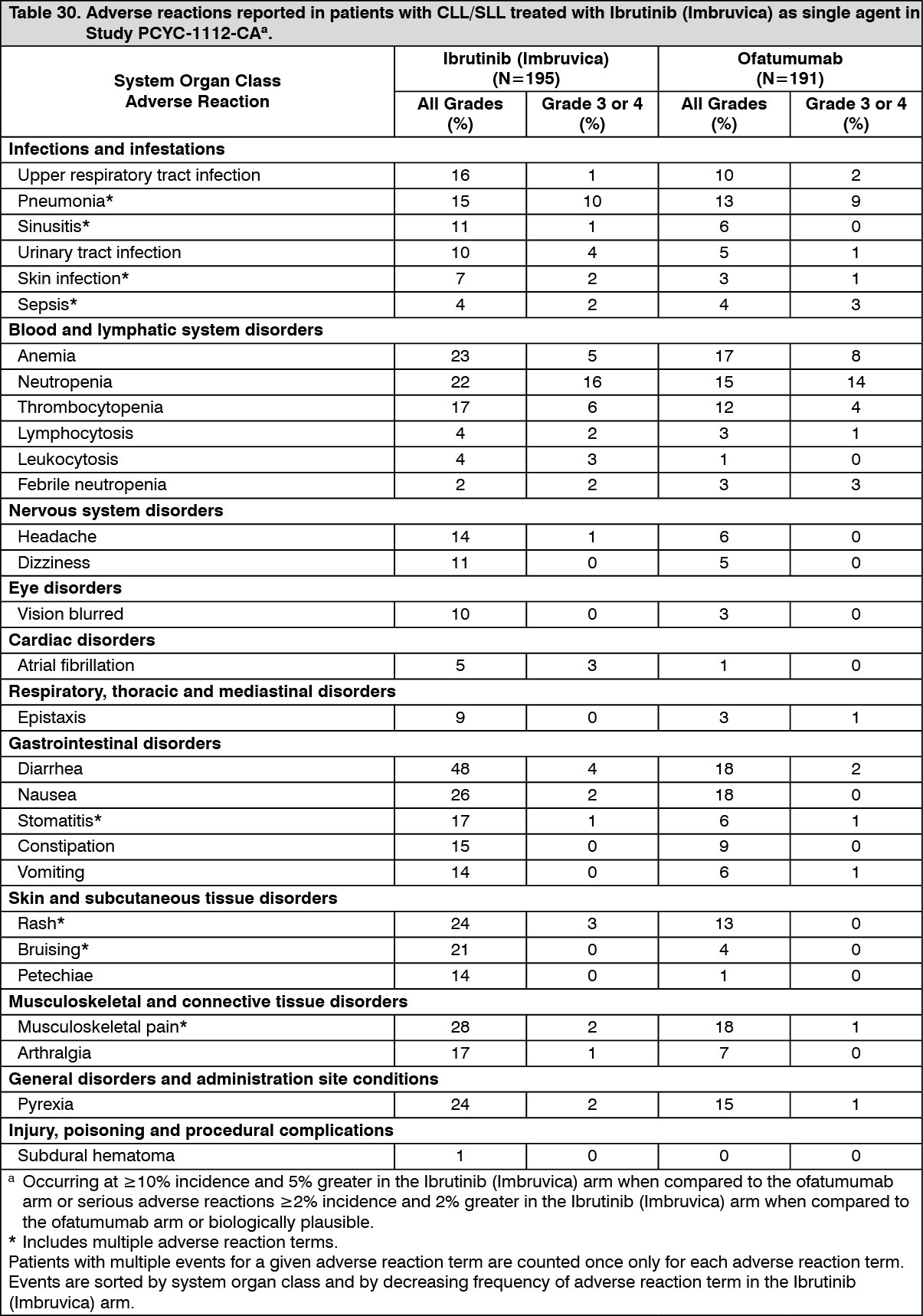 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageCombination therapy: Adverse reactions described as follows in Table 31 reflect exposure to Ibrutinib (Imbruvica) + BR with a median duration of 14.7 months and exposure to placebo + BR with a median of 12.8 months in Study CLL3001. (See Table 31.)
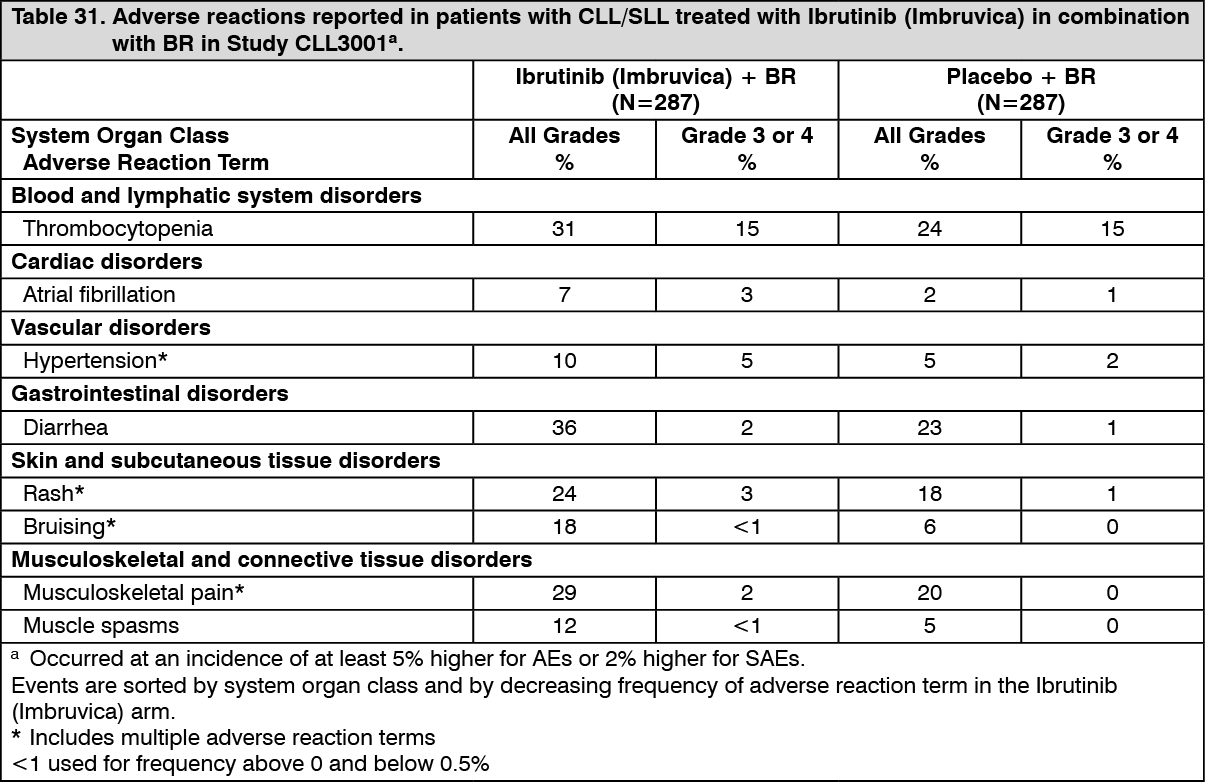 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageWaldenström's macroglobulinemia (WM): The data described as follows reflect exposure to Ibrutinib (Imbruvica) in an open‑label clinical study that included 63 patients with previously treated WM (PCYC-1118E) and a randomized phase 3 clinical study in 75 patients with treatment-naïve or previously treated WM (PCYC-1127-CA). Study PCYC-1127-CA also had an additional monotherapy arm of 31 patients with previously treated WM who failed prior rituximab-containing therapy. The safety profile of patients included in the PCYC-1127-CA monotherapy arm is consistent with the overall known WM safety profile for Ibrutinib (Imbruvica)-exposed patients.
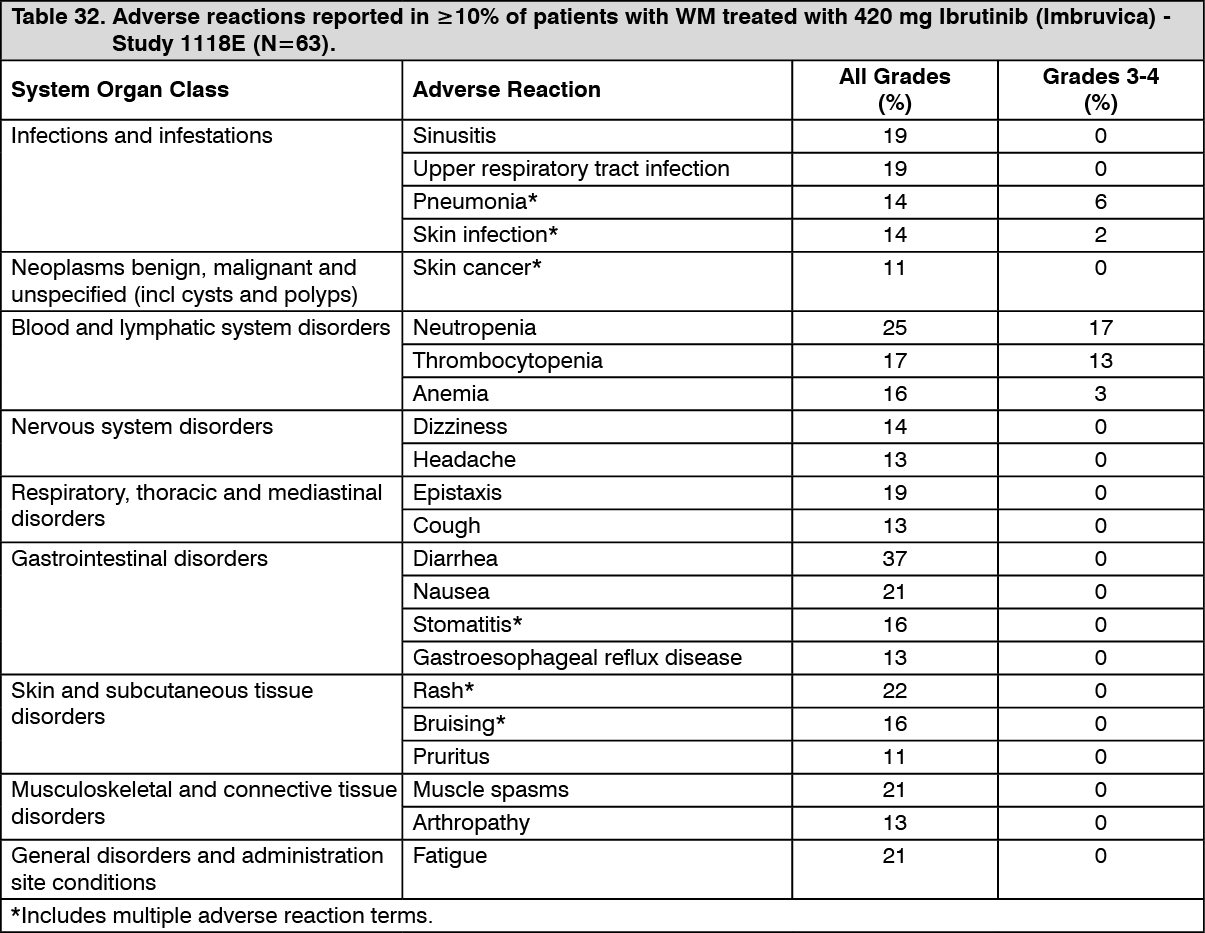
The most commonly occurring adverse reactions in the WM studies (PCYC-1118E and PCYC-1127-CA) (≥20%) were hemorrhage (e.g., bruising), diarrhea, musculoskeletal pain, rash, nausea, and neutropenia.
The most common Grade 3/4 adverse reactions (≥5%) were: neutropenia, pneumonia, hypertension, atrial fibrillation, and thrombocytopenia.
Discontinuation and dose reduction due to ARs: Four percent of patients receiving Ibrutinib (Imbruvica) in the WM studies (PCYC-1118E and PCYC-1127-CA) discontinued treatment due to adverse reactions. Adverse reactions leading to dose reduction occurred in 11% of patients.
Adverse reactions described as follows in Table 32 reflect exposure to Ibrutinib (Imbruvica) with a median duration of 11.7 months in Study PCYC-1118E. (See Table 32.)
 Click on icon to see table/diagram/image
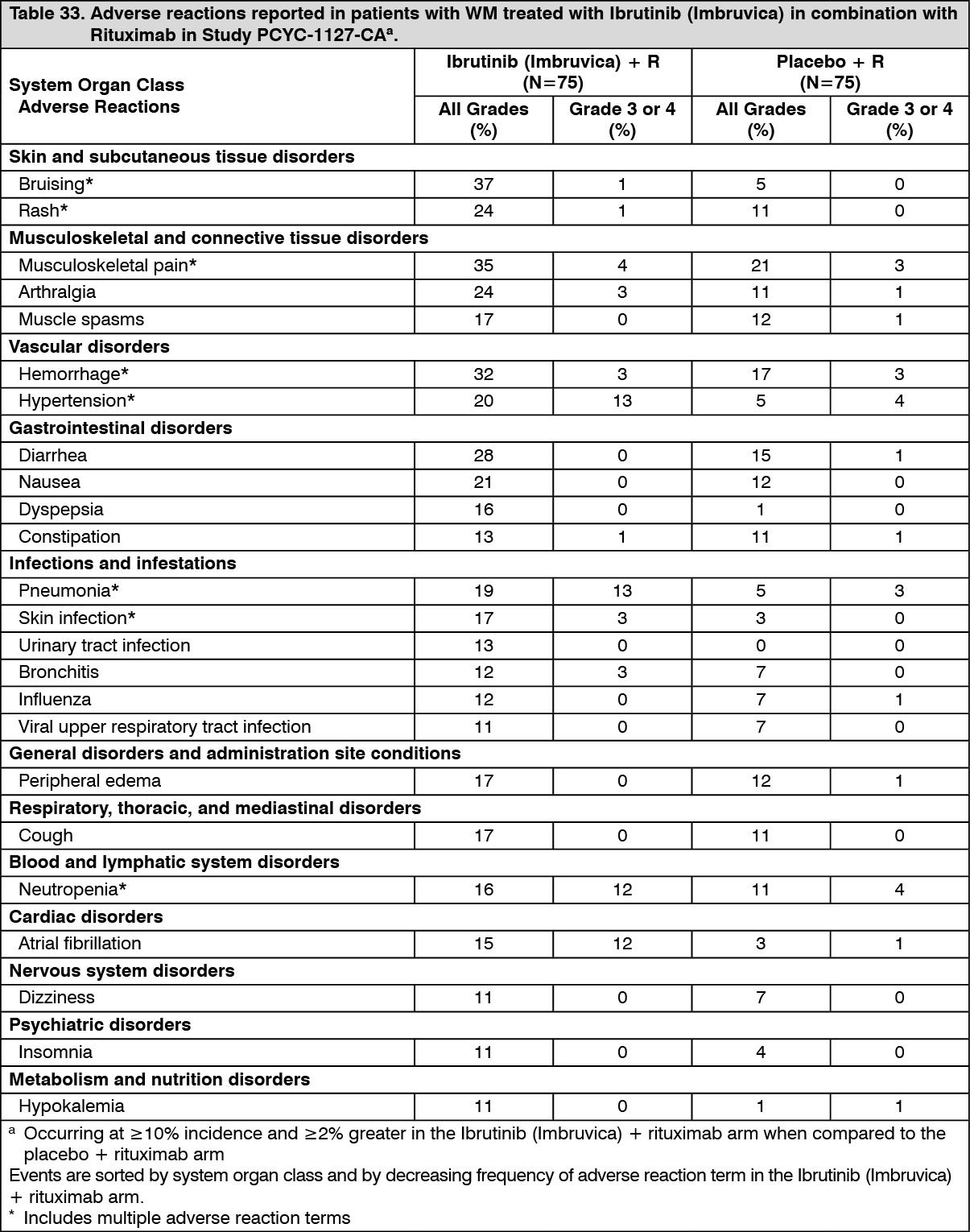
Click on icon to see table/diagram/imageAdverse reactions from Study PCYC-1127-CA are described as follows in Table 33 reflecting exposure to Ibrutinib (Imbruvica) + rituximab with a median duration of 25.8 months and exposure to placebo + rituximab with a median duration of 15.5 months in patients with treatment-naïve or previously treated WM. (See Table 33.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageGrade 3 or 4 infusion-related reactions were observed in 1% of patients treated with Ibrutinib (Imbruvica) + rituximab and 16% of patients treated with placebo + rituximab.
Long-term safety: The safety data from long-term treatment with Ibrutinib (Imbruvica) over 5 years from 1284 patients (treatment-naïve CLL/SLL n=162, relapsed/refractory CLL/SLL n=646, relapsed/refractory MCL n=370, and WM n=106) were analyzed. The median duration of treatment for CLL/SLL was 51 months (range, 0.2 to 98 months) with 70% and 52% of patients receiving treatment for more than 2 years and 4 years, respectively. The median duration of treatment for MCL was 11 months (range, 0 to 87 months) with 31% and 17% of patients receiving treatment for more than 2 years and 4 years, respectively. The median duration of treatment for WM was 47 months (range, 0.3 to 61 months) with 78% and 46% of patients receiving treatment for more than 2 years and 4 years, respectively. The overall known safety profile of Ibrutinib (Imbruvica)-exposed patients remained consistent, other than an increasing prevalence of hypertension, with no new safety concerns identified. The prevalence for Grade 3 or greater hypertension was 4% (year 0‑1), 7% (year 1-2), 9% (year 2-3), and 9% (year 3-4), and 9% (year 4-5); the overall incidence for the 5-year period was 11%.
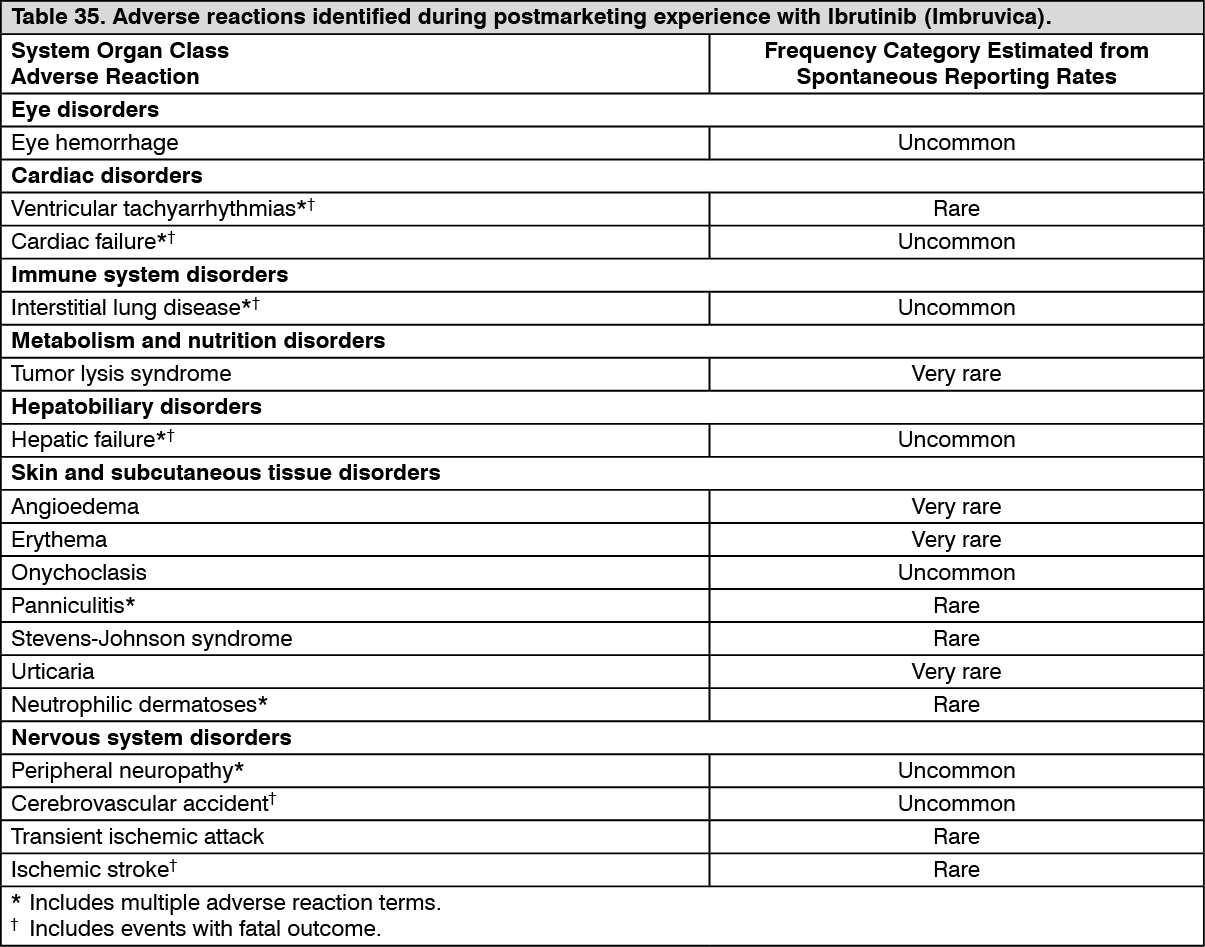
Postmarketing data: In addition to the adverse reactions reported during clinical studies and listed previously, the following adverse reactions have been reported during postmarketing experience (Table 35). Because these reactions were reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. In the table, the frequencies are provided according to the following convention: see Table 34.
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn Table 35, adverse reactions are presented by frequency category based on spontaneous reporting rates. (See Table 35.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
View ADR Monitoring Form