


 Sign Out
Sign Out

The safety of trametinib in combination with dabrafenib has been evaluated in the integrated safety population of 1076 patients with BRAF V600 mutant unresectable or metastatic melanoma, Stage III BRAF V600 mutant melanoma following complete resection (adjuvant treatment) and advanced NSCLC treated with trametinib 2 mg once daily and dabrafenib 150 mg twice daily. Of these patients, 559 were treated with the combination for BRAF V600 mutant melanoma in two randomized Phase III studies, MEK115306 (COMBI-d) and MEK116513 (COMBI-v) (see Pharmacology: Pharmacodynamics: Clinical Studies: Combination therapy under Actions), and 82 were treated with the combination for BRAF V600 mutant NSCLC in a multi cohort, non randomised Phase II study BRF113928 (see Pharmacology: Pharmacodynamics: Clinical Studies under Actions).
The most common adverse reactions (incidence ≥ 20 %) for trametinib and dabrafenib combination therapy include pyrexia, fatigue, nausea, chills, headache, diarrhoea, vomiting, arthralgia, and rash.
Tabulated summary of adverse reactions: Adverse reactions are listed as follows by MedDRA body system organ class.
The following convention has been utilized for the classification of frequency: Very common ≥1/10; Common ≥1/100 to <1/10; Uncommon ≥1/1,000 to <1/100; Rare ≥1/10,000 to <1/1,000; Not known (cannot be estimated from the available data).
Categories have been assigned based on absolute frequencies in the clinical trial data. Within each frequency grouping, adverse reactions are presented in order of decreasing seriousness. (See Tables 12, 13a and 13b.)
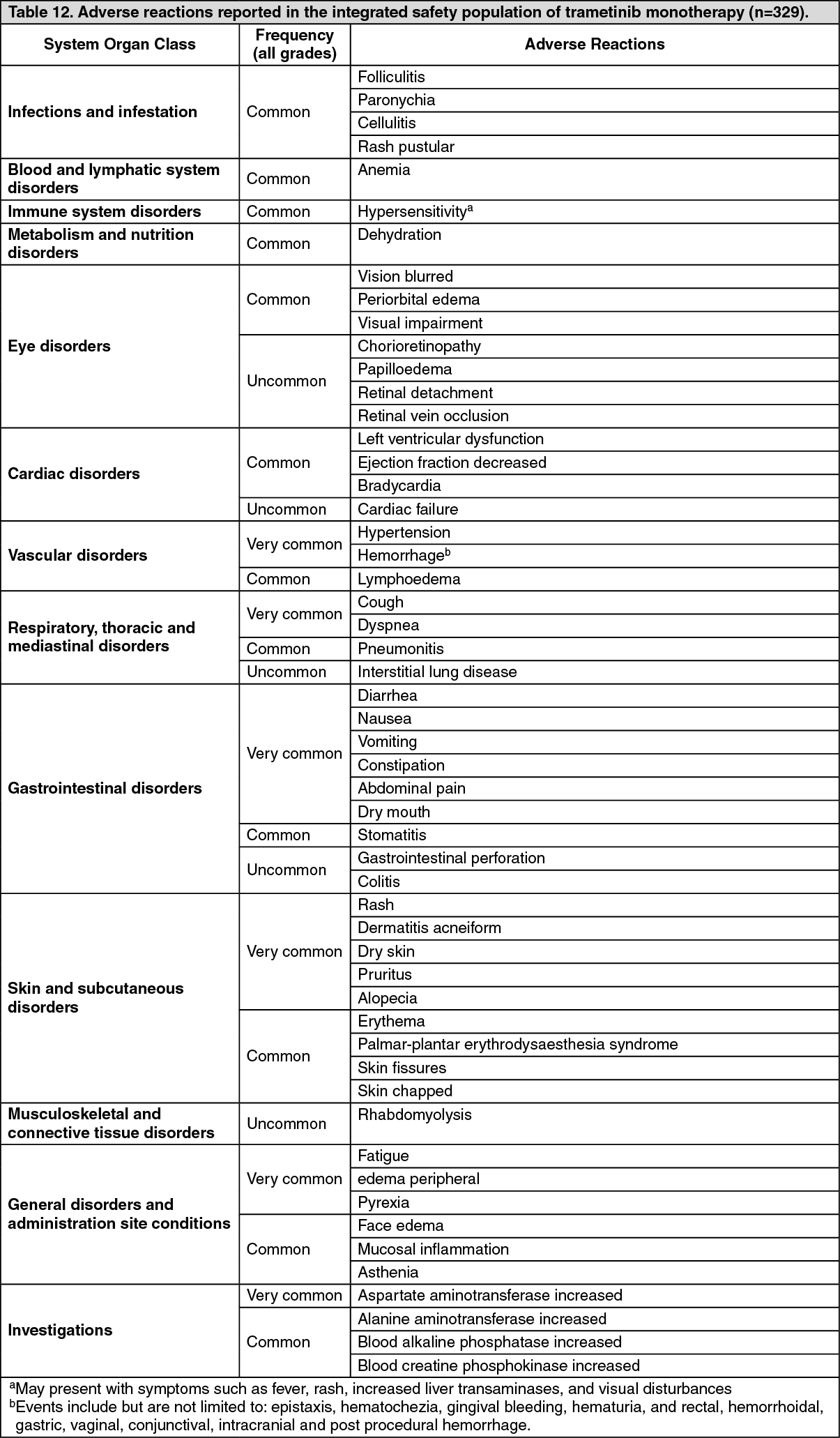 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image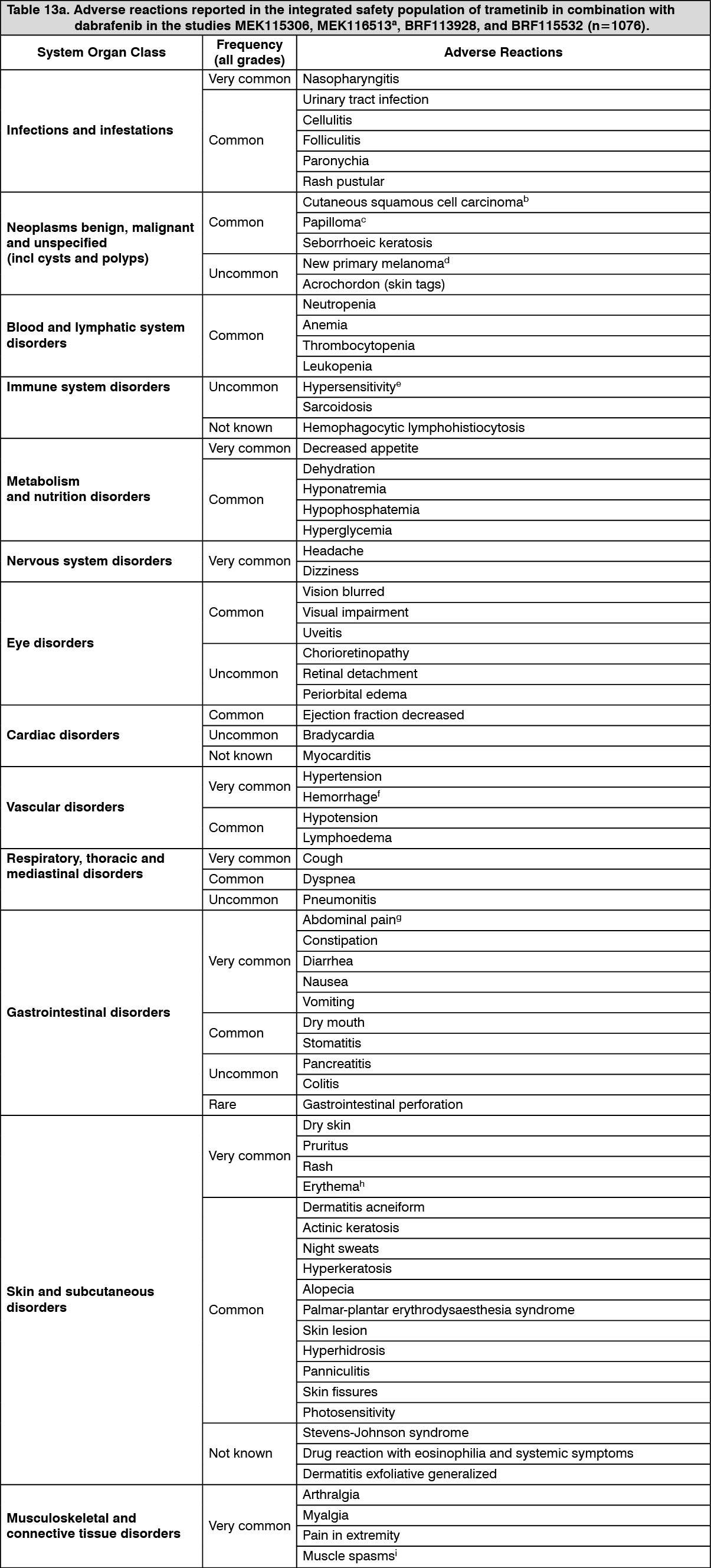 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image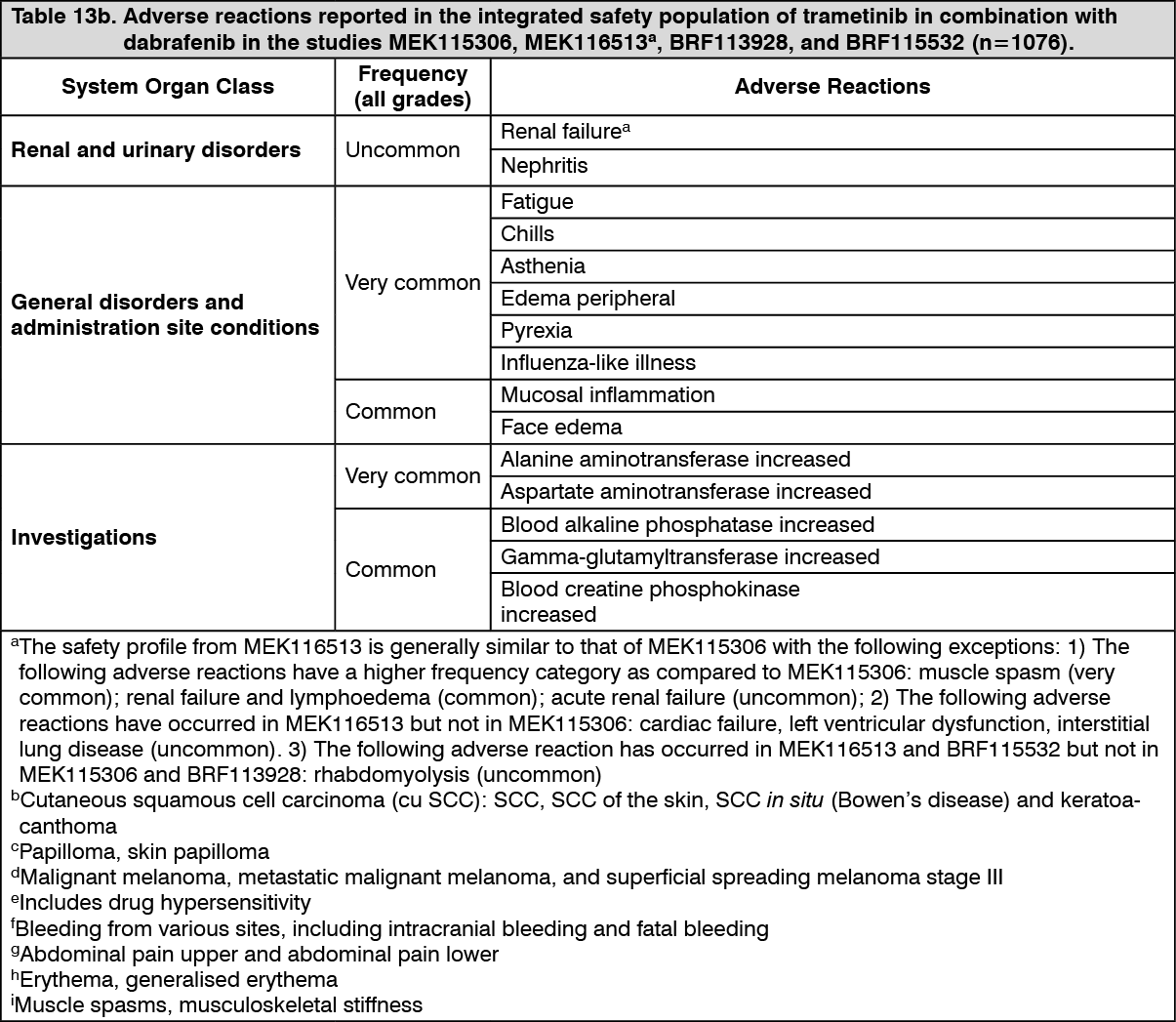 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageDescription of selected adverse reactions: New malignancies: New malignancies, cutaneous and non-cutaneous, can occur when trametinib is used in combination with dabrafenib. Please refer to the Full Prescribing Information of dabrafenib.
Hemorrhage: Hemorrhagic events, including major hemorrhagic events and fatal hemorrhages occurred in patients taking trametinib as monotherapy and in combination with dabrafenib. The majority of bleeding events were mild. Fatal intracranial hemorrhages occurred in the integrated safety population of trametinib in combination with dabrafenib in ≤1% (8/1076) of patients. The median time to onset of the first occurrence of haemorrhagic events for the combination of trametinib and dabrafenib was 94 days in the melanoma Phase III studies and 75 days in the NSCLC study for the patients who had received prior anti-cancer therapy.
The risk of hemorrhage may be increased with concomitant use of antiplatelet or anticoagulant therapy. If hemorrhage occurs, treat as clinically indicated (see Precautions).
LVEF reduction/Left ventricular dysfunction: Trametinib has been reported to decrease LVEF when used as monotherapy or in combination with dabrafenib. In clinical trials, the median time to first occurrence of left ventricular dysfunction, cardiac failure and LVEF decrease was between 2 to 5 months. In the integrated safety population of trametinib in combination with dabrafenib, decreased LVEF has been reported in 6% (65/1076) of patients with most cases being asymptomatic and reversible. Patients with LVEF lower than the institutional lower limit of normal were not included in clinical trials with trametinib. Trametinib should be used with caution in patients with conditions that could impair left ventricular function (see Dosage & Administration and Precautions).
Pyrexia: Pyrexia has been reported in clinical trials with trametinib as monotherapy and in combination with dabrafenib; however, the incidence and severity of pyrexia are increased with the combination therapy. Please refer to Precautions and Adverse Reactions of the dabrafenib Prescribing Information.
Hepatic events: Hepatic adverse events have been reported in clinical trials with trametinib as monotherapy and in combination with dabrafenib. Of the hepatic adverse reactions, increased ALT and AST were the most common events and the majority were either Grade 1 or 2. For trametinib monotherapy, more than 90% of these liver events occurred within the first 6 months of treatment. Liver events were detected in clinical trials with monitoring every four weeks. It is recommended that patients receiving treatment with trametinib monotherapy or in combination with dabrafenib have liver function monitored every four weeks for 6 months. Liver monitoring may be continued thereafter as clinically indicated (see Precautions).
Hypertension: Elevations in blood pressure have been reported in association with trametinib as monotherapy and in combination with dabrafenib, in patients with or without pre-existing hypertension. Blood pressure should be measured at baseline and monitored during treatment, with control of hypertension by standard therapy as appropriate (see Precautions).
Interstitial lung disease (ILD)/Pneumonitis: Patients treated with trametinib or combination with dabrafenib may develop ILD or pneumonitis. Trametinib should be withheld in patients with suspected ILD or pneumonitis, including patients presenting with new or progressive pulmonary symptoms and findings including cough, dyspnea, hypoxia, pleural effusion, or infiltrates, pending clinical investigations. For patients diagnosed with treatment-related ILD or pneumonitis trametinib should be permanently discontinued (see Dosage & Administration and Precautions).
Visual impairment: Disorders associated with visual disturbances, including RPED and RVO, have been observed with trametinib. Symptoms such as blurred vision, decreased acuity, and other visual disturbances have been reported in the clinical trials with trametinib (see Dosage & Administration and Precautions).
Rash: Rash has been observed in about 60% of patients when given as monotherapy and in about 24% of patients in trametinib and dabrafenib combination studies in the integrated safety population. The majority of these cases were Grade 1 or 2 and did not require any dose interruptions or dose reductions (see Dosage & Administration and Precautions).
Rhabdomyolysis: Rhabdomyolysis has been reported in patients taking trametinib alone or in combination with dabrafenib. Signs or symptoms of rhabdomyolysis should warrant an appropriate clinical evaluation and treatment as indicated (see Precautions).
Pancreatitis: Pancreatitis has been reported with dabrafenib in combination with trametinib. Please refer to the Full Prescribing Information of dabrafenib.
Renal failure: Renal failure has been reported with dabrafenib in combination with trametinib. Please refer to the Full Prescribing Information of dabrafenib.
Special populations: Elderly: In the Phase III study with trametinib in patients with unresectable or metastatic melanoma (n = 211), 49 patients (23%) were ≥65 years of age, and 9 patients (4%) were ≥75 years of age. The proportion of subjects experiencing adverse reactions (AR) and serious adverse reactions (SAR) was similar in the subjects aged <65 years and those aged ≥65 years. Patients ≥65 years were more likely to experience ARs leading to permanent discontinuation of medicinal product, dose reduction and dose interruption than those <65 years.
In the integrated safety population of trametinib in combination with dabrafenib (n = 1076) 265 patients (25%) were ≥65 years of age; 62 patients (6%) were ≥75 years of age. The proportion of patients experiencing ARs was similar in those aged <65 years and those aged ≥65 years in all studies. Patients ≥65 years were more likely to experience SARs and ARs leading to permanent discontinuation of medicinal product, dose reduction and dose interruption than those <65 years.
Renal impairment: No dosage adjustment is required in patients with mild or moderate renal impairment (see Pharmacology: Pharmacokinetics under Actions). Trametinib should be used with caution in patients with severe renal impairment (see Dosage & Administration and Precautions).
Hepatic impairment: No dosage adjustment is required in patients with mild hepatic impairment (see Pharmacology: Pharmacokinetics under Actions). Trametinib should be used with caution in patients with moderate or severe hepatic impairment (see Dosage & Administration and Precautions).
Trametinib in combination with dabrafenib in patients with brain metastases: The safety and efficacy of the combination of trametinib and dabrafenib have been evaluated in a multi-cohort, open-label, Phase II study in patients with BRAF V600 mutant melanoma with brain metastases. The safety profile observed in these patients appears to be consistent with the integrated safety profile of the combination.
Reporting of suspected adverse reactions: Reporting suspected adverse reactions after authorization of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product.
View ADR Monitoring Form