Pharmacotherapeutic group: Antineoplastic agents, protein kinase inhibitor, Mitogen activated protein kinase (MEK) inhibitors.
ATC code: L01EE01.
Pharmacology: Pharmacodynamics: Mechanism of action: Trametinib is a reversible, highly selective, allosteric inhibitor of mitogen-activated extracellular signal regulated kinase 1 (MEK1) and MEK2 activation and kinase activity. MEK proteins are components of the extracellular signal-related kinase (ERK) pathway. In melanoma and other cancers, this pathway is often activated by mutated forms of BRAF which activates MEK. Trametinib inhibits activation of MEK by BRAF and inhibits MEK kinase activity. Trametinib inhibits growth of BRAF V600 mutant melanoma cell lines and demonstrates anti-tumor effects in BRAF V600 mutant melanoma animal models.
Combination with dabrafenib: Dabrafenib is an inhibitor of RAF kinases. Oncogenic mutations in BRAF lead to constitutive activation of the RAS/RAF/MEK/ERK pathway. Thus, trametinib and dabrafenib inhibit two kinases in this pathway, MEK and RAF, and therefore the combination provides concomitant inhibition of the pathway. The combination of trametinib with dabrafenib has shown anti-tumor activity in BRAF V600 mutation positive melanoma cell lines
in vitro and delays the emergence of resistance
in vivo in BRAF V600 mutation positive melanoma xenografts.
Determination of BRAF mutation status: Before taking trametinib or the combination with dabrafenib, patients must have BRAF V600 mutation-positive tumor status confirmed by a validated test.
In clinical trials, central testing for BRAF V600 mutation using a BRAF mutation assay was conducted on the most recent tumor sample available. Primary tumor or tumor from a metastatic site was tested with a validated polymerase chain reaction (PCR) assay developed by Response Genetics Inc. The assay was specifically designed to differentiate between the V600E and V600K mutations. Only patients with BRAF V600E or V600K mutation positive tumors were eligible for study participation.
Subsequently, all patient samples were re-tested using the CE-marked bioMerieux (bMx) THxID BRAF validated assay. The bMx THxID BRAF assay is an allele-specific PCR performed on DNA extracted from FFPE tumor tissue. The assay was designed to detect the BRAF V600E and V600K mutations with high sensitivity (down to 5% V600E and V600K sequence in a background of wild-type sequence using DNA extracted from FFPE tissue). Non-clinical
and clinical trials with retrospective bi-directional Sanger sequencing analyses have shown that the test also detects the less common BRAF V600D mutation and V600E/K601E mutation with lower sensitivity. Of the specimens from the non-clinical and clinical trials (n = 876) that were mutation positive by the THxID BRAF assay and subsequently were sequenced using the reference method, the specificity of the assay was 94%.
Pharmacodynamic effects: Trametinib suppressed levels of phosphorylated ERK in BRAF mutant melanoma tumor cell lines and melanoma xenograft models.
In patients with BRAF and NRAS mutation positive melanoma, administration of trametinib resulted in dose-dependent changes in tumor biomarkers including inhibition of phosphorylated ERK, inhibition of Ki67 (a marker of cell proliferation), and increases in p27 (a marker of apoptosis). The mean trametinib concentrations observed following repeat dose administration of 2 mg once daily exceeds the preclinical target concentration over the 24-hr
dosing interval, thereby providing sustained inhibition of the MEK pathway.
Clinical Studies: Unresectable or metastatic melanoma: In the clinical trials only patients with cutaneous melanoma were studied. Efficacy in patients with ocular or mucosal melanoma has not been assessed.
Trametinib in combination with dabrafenib: Treatment naïve patients: The efficacy and safety of the recommended dose of trametinib (2 mg once daily) in combination with dabrafenib (150 mg twice daily) for the treatment of adult patients with unresectable or metastatic melanoma with a BRAF V600
mutation was studied in two Phase III studies and one supportive Phase I/II study.
MEK115306 (COMBI-d): MEK115306 was a Phase III, randomized, double-blinded study comparing the combination of dabrafenib and trametinib to dabrafenib and placebo in first-line therapy for subjects with unresectable (Stage IIIC) or metastatic (Stage IV) BRAF V600E/K mutation-positive cutaneous melanoma. The primary endpoint of the study was progression-free survival (PFS), with a key secondary endpoint of overall survival (OS). Subjects were stratified by lactate dehydrogenase (LDH) level (> the upper limit of normal (ULN) versus ≤ULN) and BRAF mutation (V600E versus V600K).
A total of 423 subjects were randomized 1:1 to either combination (N = 211) or dabrafenib (N = 212). Most subjects were Caucasian (>99%) and male (53%), with a median age of 56 years (28% were ≥65 years). The majority of subjects had Stage IVM1c disease (67%). Most subjects had LDH ≤ULN (65%), Eastern Cooperative Oncology Group (ECOG) performance status of 0 (72%), and visceral disease (73%) at baseline. The majority of subjects had a BRAF V600E mutation (85%). Subjects with brain metastases were not included in the trial.
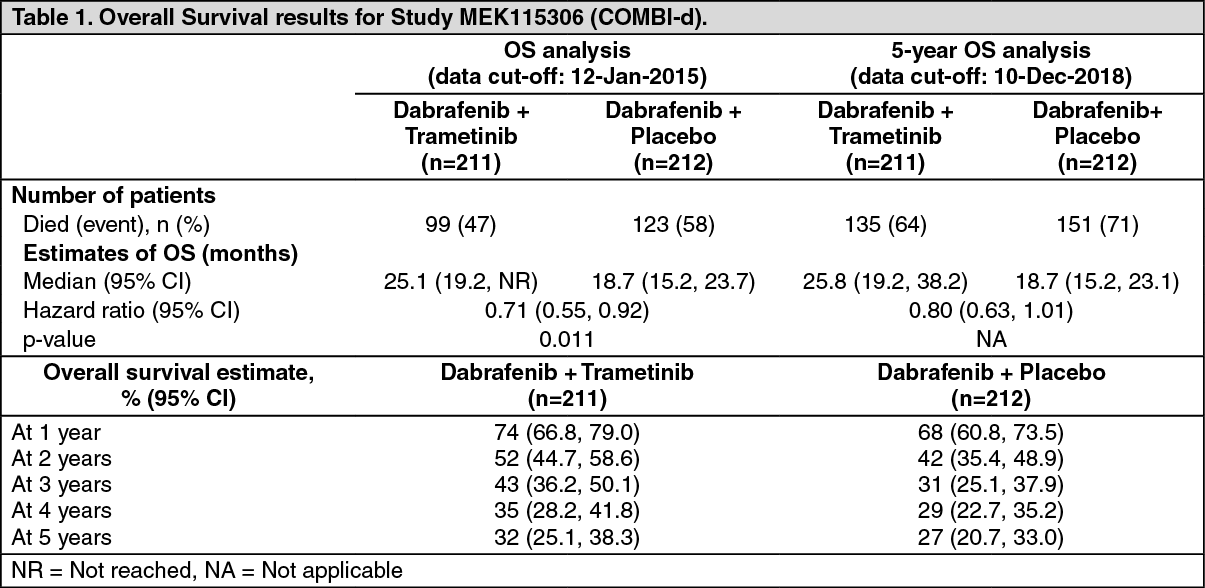
Median OS and estimated 1 year, 2 year, 3 year, 4 year and 5 year survival rates are presented in Table 1. From an OS analysis at 5 years, the median OS for the combination arm was approximately 7 months longer than for dabrafenib monotherapy (25.8 months versus 18.7 months) with 5-year survival rates of 32% for the combination versus 27% for dabrafenib monotherapy (Table 1, Figure 1). The Kaplan-Meier OS curve appears to stabilise
from 3 to 5 years (see Figure 1). The 5-year overall survival rate was 40% (95% CI: 31.2, 48.4) in the combination arm versus 33% (95% CI: 25.0, 41.0) in the dabrafenib monotherapy arm for patients who had a normal lactate dehydrogenase level at baseline, and 16% (95% CI: 8.4, 26.0) in the combination arm versus 14% (95% CI: 6.8, 23.1) in the dabrafenib monotherapy arm for patients with an elevated lactate dehydrogenase level at baseline. (See Table 1 and Figure 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Improvements for the primary endpoint of PFS were sustained over a 5 year timeframe in the combination arm compared to dabrafenib monotherapy. Improvements were also observed for overall response rate (ORR) and a longer duration of response (DoR) was observed in the combination arm compared to dabrafenib monotherapy (Table 2).
Click on icon to see table/diagram/image
MEK116513 (COMBI-v): Study MEK116513 was a 2-arm, randomized, open-label, Phase III study comparing dabrafenib and trametinib combination therapy with vemurafenib monotherapy in BRAF V600 mutation-positive metastatic unresectable or melanoma. The primary endpoint of the study was overall survival with a key secondary endpoint of PFS. Subjects were stratified by lactate dehydrogenase (LDH) level (> the upper limit of normal (ULN) versus ≤ ULN) and BRAF mutation (V600E versus V600K).
A total of 704 subjects were randomized 1:1 to either combination or vemurafenib. Most subjects were Caucasian (>96%) and male (55%), with a median age of 55 years (24% were ≥65 years). The majority of subjects had Stage IV M1c disease (61% overall). Most subjects had LDH ≤ULN (67%), ECOG performance status of 0 (70%), and visceral disease (78%) at baseline. Overall, 54% of subjects had <3 disease sites at baseline. The majority of subjects had BRAF V600E mutation-positive melanoma (89%). Subjects with brain metastases were not included in the trial.
Median OS and estimated 1-year, 2-year, 3-year, 4-year and 5-year survival rates are presented in Table 3. From an OS analysis at 5 years, the median OS for the combination arm was approximately 8 months longer than the median OS for vemurafenib monotherapy (26.0 months versus 17.8 months) with 5-year survival rates of 36% for the combination versus 23% for vemurafenib monotherapy (Table 3, Figure 2). The Kaplan-Meier OS curve appears to stabilise from 3 to 5 years (see Figure 2). The 5-year overall survival rate was 46% (95% CI: 38.8, 52.0) in the combination arm versus 28% (95% CI: 22.5, 34.6) in the vemurafenib monotherapy arm for patients who had a normal lactate dehydrogenase level at baseline, and 16% (95% CI: 9.3, 23.3) in the combination arm versus 10% (95% CI: 5.1, 17.4) in the vemurafenib monotherapy arm for patients with an elevated lactate dehydrogenase level at baseline. (See Table 3 and Figure 2.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Improvements for the secondary endpoint of PFS were sustained over a 5 year timeframe in the combination arm compared to vemurafenib monotherapy. Improvements were also observed for ORR and a longer DoR was observed in the combination arm compared to vemurafenib monotherapy (Table 4).
Click on icon to see table/diagram/image
Prior BRAF inhibitor therapy: There are limited data in patients taking the combination of trametinib with dabrafenib who have progressed on a
prior BRAF inhibitor.
Part B of study BRF113220 included a cohort of 26 patients that had progressed on a BRAF inhibitor. The trametinib 2 mg once daily and dabrafenib 150 mg twice daily combination demonstrated limited clinical activity in patients who had progressed on a BRAF inhibitor (see Precautions). The investigator-assessed confirmed response rate was 15% (95% CI: 4.4, 34.9) and the median PFS was 3.6 months (95% CI: 1.9, 5.2). Similar results were seen in the 45 patients who crossed over from dabrafenib monotherapy to the trametinib 2 mg once daily and dabrafenib 150 mg twice daily combination in Part C of this study. In these patients a 13% (95% CI: 5.0, 27.0) confirmed response rate was observed with a median PFS of 3.6 months (95% CI: 2, 4).
Patients with brain metastases: The efficacy and safety of trametinib in combination with dabrafenib in patients with BRAF mutant-positive melanoma that has metastasized to the brain was studied in a non-randomized, open-label, multicentre, Phase II study (COMBI-MB study). A total of 125 patients were enrolled into four cohorts: Cohort A: patients with BRAFV600E mutant melanoma with asymptomatic brain metastases without prior local brain-directed therapy and ECOG performance status of 0 or 1.
Cohort B: patients with BRAFV600E mutant melanoma with asymptomatic brain metastases with prior local brain-directed therapy and ECOG performance status of 0 or 1.
Cohort C: patients with BRAFV600D/K/R mutant melanoma with asymptomatic brain metastases, with or without prior local brain-directed therapy and ECOG performance status of 0 or 1.
Cohort D: patients with BRAFV600D/E/K/R mutant melanoma with symptomatic brain metastases, with or without prior local brain-directed therapy and ECOG performance status of 0 or 1 or 2.
The primary endpoint of the study was intracranial response in Cohort A, defined as the percentage of patients with a confirmed intracranial response assessed by the investigator using modified Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1. Intracranial response assessed by the
investigator in Cohorts B, C and D were secondary endpoints of the study. Due to small sample size reflected by wide 95% CIs, the results in Cohorts B, C, and D should be interpreted with caution. Efficacy results are summarised in Table 3. (See Table 5.)
Click on icon to see table/diagram/image
Trametinib monotherapy: Treatment naïve patients: The efficacy and safety of trametinib in patients with BRAF unresectable or metastatic mutant melanoma (V600E and V600K) were evaluated in a randomized open-label Phase III study (MEK114267[METRIC]). Measurement of patients' BRAF V600 mutation status was required.
Patients (N = 322) who were treatment naïve or may have received one prior chemotherapy treatment in the metastatic setting [Intent to Treat (ITT) population] were randomized 2:1 to receive trametinib 2 mg once daily or chemotherapy (dacarbazine 1000 mg/m
2 every 3 weeks or paclitaxel 175 mg/m
2 every 3 weeks). Treatment for all patients continued until disease progression, death or withdrawal.
The primary endpoint of the study was to evaluate the efficacy of trametinib compared to chemotherapy with respect to PFS in patients with advanced/metastatic BRAF V600E/K mutation-positive melanoma without a prior history of brain metastases (N = 273) which is considered the primary efficacy population. The secondary endpoints were PFS in the ITT population and OS, ORR, and DoR in the primary efficacy population and ITT population. Patients in the chemotherapy arm were allowed to cross-over to the trametinib arm after independent confirmation of progression. Of the patients with confirmed disease progression in the chemotherapy arm, a total of 51 (47%) crossed over to receive trametinib.
Baseline characteristics were balanced between treatment groups in the primary efficacy population and the ITT population. In the ITT population, 54% of patients were male and all were Caucasian. The median age was 54 years (22% were ≥65 years); all patients had an ECOG performance score of 0 or 1; and 3% had history of brain metastases. Most patients (87%) in the ITT population had BRAF V600E mutation and 12% of patients had BRAF V600K. Most patients (66%) received no prior chemotherapy for advanced or metastatic disease.
The efficacy results in the primary efficacy population were consistent with those in the ITT population; therefore, only the efficacy data for the ITT population are presented in Table 3. Kaplan-Meier curves of investigator assessed OS (post-hoc analysis 20 May 2013) is presented in Figure 3. (See Table 6.)
Click on icon to see table/diagram/image
The PFS result was consistent in the subgroup of patients with V600K mutation positive melanoma (HR = 0.50; [95% CI: 0.18, 1.35], p=0.0788).
An additional OS analysis was undertaken based upon the 20 May 2013 data cut, see Table 4.
For October 2011, 47% of subjects had crossed over, while for May 2013, 65% of subjects had crossed over. (See Table 7 and Figure 3.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Prior BRAF inhibitor therapy: In a single-arm Phase II study, designed to evaluate the objective response rate, safety, and pharmacokinetics following dosing of trametinib at 2 mg once daily in patients with BRAF V600E, V600K, or V600D mutation-positive metastatic melanoma (MEK113583), two separate cohorts were enrolled: Cohort A: patients with prior treatment with a BRAF inhibitor either with or without other prior therapy, Cohort B: patients with at least 1 prior chemotherapy or immunotherapy, without prior treatment with a BRAF inhibitor.
In Cohort A of this study, trametinib did not demonstrate clinical activity in patients who had progressed on a prior BRAF inhibitor therapy.
Non-small cell lung cancer: Study BRF113928: The efficacy and safety of trametinib in combination with dabrafenib was studied in a Phase II,
three-cohort, multicenter, non-randomized and open-label study in which patients with Stage IV BRAF V600E mutant NSCLC were enrolled. The primary endpoint was ORR using the RECIST 1.1 assessed by the investigator. Secondary endpoints included DoR, PFS, OS, safety and population pharmacokinetics. ORR, DoR and PFS were also assessed by an Independent Review Committee (IRC) as a sensitivity analysis.
Cohorts were enrolled sequentially: Cohort A: Monotherapy (dabrafenib 150 mg twice daily), 84 patients enrolled. 78 patients had previous systemic treatment for their metastatic disease.
Cohort B: Combination therapy (dabrafenib 150 mg twice daily and trametinib 2 mg once daily), 59 patients enrolled. 57 patients had 1-3 lines of previous systemic treatment for their metastatic disease. 2 patients had no previous systemic treatment and were included in the analysis for patients enrolled in Cohort C.
Cohort C: Combination therapy (dabrafenib 150 mg twice daily and trametinib 2 mg once daily), 34 patients. All patients received study medicinal product as first-line treatment for metastatic disease.
Among the total of 93 patients who were enrolled in the combination therapy cohorts B and C, most patients were Caucasian (>90%), and similar female versus male (54% versus 46%), with a median age of 64 years in second-line or higher patients and 68 years in the first-line patients. Most patients (94%)
enrolled in the combination-therapy-treated cohorts had an ECOG performance status of 0 or 1. 26 (28%) had never smoked. The majority of patients had a non-squamous histology. In the previously-treated population, 38 patients (67%) had one line of systemic anti-cancer therapy for metastatic disease.
At the time of the primary analysis, the primary endpoint of investigator-assessed ORR in the first-line population was 61.1% (95% CI, 43.5%, 76.9%) and in the previously-treated population was 66.7% (95% CI, 52.9%, 78.6%). These met the statistical significance to reject the null hypothesis that the ORR of dabrafenib in combination with trametinib for this NSCLC population was less than or equal to 30%. The final analysis of efficacy performed 5 years after last subject first dose is presented in Table 8. (See Table 8.)
Click on icon to see table/diagram/image
Other studies - pyrexia management analysis: Study CPDR001F2301 (COMBI-i): Pyrexia is observed in patients treated with dabrafenib and trametinib combination therapy. The initial registration studies for the combination therapy in the unresectable or metastatic melanoma setting (COMBI-d and COMBI-v; total N=559) and in the adjuvant melanoma setting (COMBI-AD, N=435) recommended to interrupt only dabrafenib in case of pyrexia (fever ≥38.5°C). In two subsequent studies in unresectable or metastatic melanoma (COMBI-i control arm, N=264) and in the adjuvant melanoma setting (COMBI-Aplus, N=552), interruption of both medicinal products when patient's temperature is ≥38°C (COMBI-Aplus), or at the first symptom of pyrexia (COMBI-i; COMBI-Aplus for recurrent pyrexia) was advised. In COMBI-i and COMBI-Aplus there was a lower incidence of grade 3/4 pyrexia, complicated pyrexia, hospitalization due to serious pyrexia adverse events of special interest (AESIs), the time spent in pyrexia AESIs, and permanent discontinuations from both medicinal products due to pyrexia AESIs (the latter in the adjuvant setting only) compared to COMBI-d, COMBI-v and COMBI-AD. The COMBI-Aplus study met its primary endpoint with a composite rate of 8.0% (95% CI: 5.9, 10.6) for grade 3/4 pyrexia, hospitalization due to pyrexia, or permanent treatment discontinuation due to pyrexia compared to 20.0% (95% CI: 16.3, 24.1) for the historical control (COMBI-AD).
Pediatric population: The safety and efficacy of trametinib as a single agent or in combination with dabrafenib have not been established in pediatric patients (see Pediatric Population under Dosage & Administration).
Pharmacokinetics: Absorption: Trametinib is absorbed orally with median time to achieve peak concentrations of 1.5 hours post-dose. The mean
absolute bioavailability of a single 2 mg tablet dose is 72% relative to an intravenous (IV) microdose. The increase in exposure (C
max and AUC) was dose-proportional following repeat dosing. Following administration of 2 mg once daily, steady-state geometric mean C
max, AUC
(0-τ) and predose concentration were 22.2 ng/ml, 370 ng*hr/ml and 12.1 ng/ml, respectively with a low peak:trough ratio (1.8). Inter-subject variability at steady state was low (<28%).
Trametinib accumulates with repeat daily dosing with a mean accumulation ratio of 6.0 at 2 mg once daily dose. Steady-state was achieved by Day 15.
Administration of a single dose of trametinib with a high-fat, high-calorie meal resulted in a 70% and 10% decrease in C
max and AUC, respectively compared to fasted conditions (see Dosage & Administration and Interactions).
Distribution: Binding of trametinib to human plasma proteins is 97.4%. Trametinib has a volume of distribution of approximately 1200 L determined following administration of a 5 μg intravenous microdose.
Biotransformation: In vitro and
in vivo studies demonstrated that trametinib is metabolized predominantly via deacetylation alone or in combination with mono-oxygenation. The deacetylated metabolite was further metabolized by glucuronidation. CYP3A4 oxidation is considered a minor pathway of metabolism. The deacetylation is mediated by the carboxyl-esterases 1b,1c and 2, with possible contribution by other hydrolytic enzymes.
Following single and repeated doses of trametinib, trametinib as parent is the main circulating component in plasma.
Elimination: Mean terminal half-life is 127 hours (5.3 days) after single dose administration. Trametinib plasma IV clearance is 3.21 L/hr.
Total dose recovery was low after a 10-day collection period (<50%) following administration of a single oral dose of radiolabelled trametinib as a solution, due to the long elimination half-life.
Drug related material was excreted predominantly in the feces (>80% of recovered radioactivity) and to a minor extent in urine (≤19%). Less than 0.1% of the excreted dose was recovered as parent in urine.
Special patient populations: Hepatic impairment: A population pharmacokinetic analysis indicates that mildly elevated bilirubin and/or AST levels (based on National Cancer Institute [NCI] classification) do not significantly affect trametinib oral clearance. No data are available in patients with moderate or severe hepatic impairment. As metabolism and biliary excretion are the primary routes of elimination of trametinib, administration of trametinib should be undertaken with caution in patients with moderate to severe hepatic impairment (see Dosage & Administration).
Renal impairment: Renal impairment is unlikely to have a clinically relevant effect on trametinib pharmacokinetics given the low renal excretion of trametinib. The pharmacokinetics of trametinib were characterized in 223 patients enrolled in clinical trials with trametinib who had mild renal impairment and 35 patients with moderate renal impairment using a population pharmacokinetic analysis. Mild and moderate renal impairment had no effect on trametinib exposure (<6% for either group). No data are available in patients with severe renal impairment (see Dosage & Administration).
Elderly: Based on the population pharmacokinetics analysis (range 19 to 92 years), age had no relevant clinical effect on trametinib pharmacokinetics. Safety data in patients ≥75 years is limited (see Adverse Reactions).
Race: There are insufficient data to evaluate the potential effect of race on trametinib pharmacokinetics as clinical experience is limited to Caucasians.
Pediatric population: No studies have been conducted to investigate the pharmacokinetics of trametinib in pediatric patients.
Body weight and gender: Based on a population pharmacokinetic analysis, gender and body weight were found to influence trametinib oral clearance. Although smaller female subjects are predicted to have higher exposure than heavier male subjects, these differences are unlikely to be clinically relevant and no dosage adjustment is warranted.
Medicinal product interactions: Effects of trametinib on drug-metabolizing enzymes and transporters:
In vitro and
in vivo data suggest that trametinib
is unlikely to affect the pharmacokinetics of other medicinal products. Based on
in vitro studies, trametinib is not an inhibitor of CYP1A2, CYP2A6, CYP2B6, CYP2D6 and CYP3A4. Trametinib was found to be an
in vitro inhibitor of CYP2C8, CYP2C9 and CYP2C19, an inducer of CYP3A4 and an inhibitor of the transporters OAT1, OAT3, OCT2, MATE1, OATP1B1, OATP1B3, Pgp and BCRP. However, based on the low dose and low clinical systemic exposure relative to the
in vitro potency of inhibition or induction values, trametinib is not considered to be an
in vivo inhibitor or inducer of these enzymes or transporters, although transient inhibition of BCRP substrates in the gut may occur (see Interactions).
Effects of other drugs on trametinib:
In vivo and
in vitro data suggest that the pharmacokinetics of trametinib are unlikely to be affected by other medicinal products. Trametinib is not a substrate of CYP enzymes or of the transporters BCRP, OATP1B1, OATP1B3, OATP2B1, OCT1, MRP2, and MATE1. Trametinib is an
in vitro substrate of BSEP and the efflux transporter P-gp. Although trametinib exposure is unlikely to be affected by inhibition of BSEP, increased levels of trametinib upon strong inhibition of hepatic P-gp cannot be excluded (see Interactions).
Effects of trametinib on other medicinal products: the effect of repeat-dose trametinib on the steady state pharmacokinetics of combination oral contraceptives, norethindrone and ethinyl estradiol, was assessed in a clinical study that consisted of 19 female patients with solid tumors. Norethindrone exposure increased by 20% and ethinyl estradiol exposure was similar when co-administered with trametinib. Based on these results, no loss of efficacy of hormonal contraceptives is expected when co administered with trametinib monotherapy.
Toxicology: Non-Clinical Safety Data: Carcinogenicity studies with trametinib have not been conducted. Trametinib was not genotoxic in studies evaluating
reverse mutations in bacteria, chromosomal aberrations in mammalian cells and micronuclei in the bone marrow of rats.
Trametinib may impair female fertility in humans, as in repeat-dose studies, increases in cystic follicles and decreases in corpora lutea were observed in female rats at exposures below the human clinical exposure based on AUC.
Additionally, in juvenile rats given trametinib, decreased ovarian weights, slight delays in hallmarks of female sexual maturation (vaginal opening and increased incidence of prominent terminal end buds within the mammary gland) and slight hypertrophy of the surface epithelium of the uterus were observed. All of these effects were reversible following an off-treatment period and attributable to pharmacology. However, in rat and dog toxicity studies up to 13 weeks in duration, there were no treatment effects observed in male reproductive tissues.
In embryo-fetal developmental toxicity studies in rats and rabbits, trametinib induced maternal and developmental toxicity. In rats decreased fetal weights and increased post-implantation loss were seen at exposures below or slightly above the clinical exposures based on AUC. In an embryo-foetal developmental toxicity study with rabbits, decreased fetal body weight, increased abortions, increased incidence of incomplete ossification and skeletal malformations were seen at sub-clinical exposures based on AUC).
In repeat-dose studies the effects seen after trametinib exposure are found mainly in the skin, gastrointestinal tract, hematological system, bone and liver. Most of the findings are reversible after drug-free recovery. In rats, hepatocellular necrosis and transaminase elevations were seen after 8 weeks at ≥0.062 mg/kg/day (approximately 0.8 times human clinical exposure based on AUC).
In mice, lower heart rate, heart weight and left ventricular function were observed without cardiac histopathology after 3 weeks at ≥0.25 mg/kg/day trametinib (approximately 3 times human clinical exposure based on AUC) for up to 3 weeks. In adult rats, mineralization of multiple organs was associated with increased serum phosphorus and was closely associated with necrosis in heart, liver and kidney and hemorrhage in the lung at exposures comparable to the human clinical exposure. In rats, hypertrophy of the physis and increased bone turnover were observed, but the physeal hypertrophy is not expected to be clinically relevant for adult humans. In rats and dogs given trametinib at or below clinical exposures, bone marrow necrosis, lymphoid atrophy in thymus and GALT and lymphoid necrosis in lymph nodes, spleen and thymus were observed, which have the potential to impair immune function. In juvenile rats, increased heart weight with no histopathology was observed at 0.35 mg/kg/day (approximately twice the adult human clinical exposure based on AUC).
Trametinib was phototoxic in an
in vitro mouse fibroblast 3T3 Neutral Red Uptake (NRU) assay at significantly higher concentrations than clinical exposures (IC50 at 2.92 μg/ml, ≥130 times the clinical exposure based on Cmax), indicating that there is low risk for phototoxicity to patients taking trametinib.
Combination with dabrafenib: In a study in dogs in which trametinib and dabrafenib were given in combination for 4 weeks, signs of gastro-intestinal
toxicity and decreased lymphoid cellularity of the thymus were observed at lower exposures than in dogs given trametinib alone. Otherwise, similar toxicities were observed as in comparable monotherapy studies.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image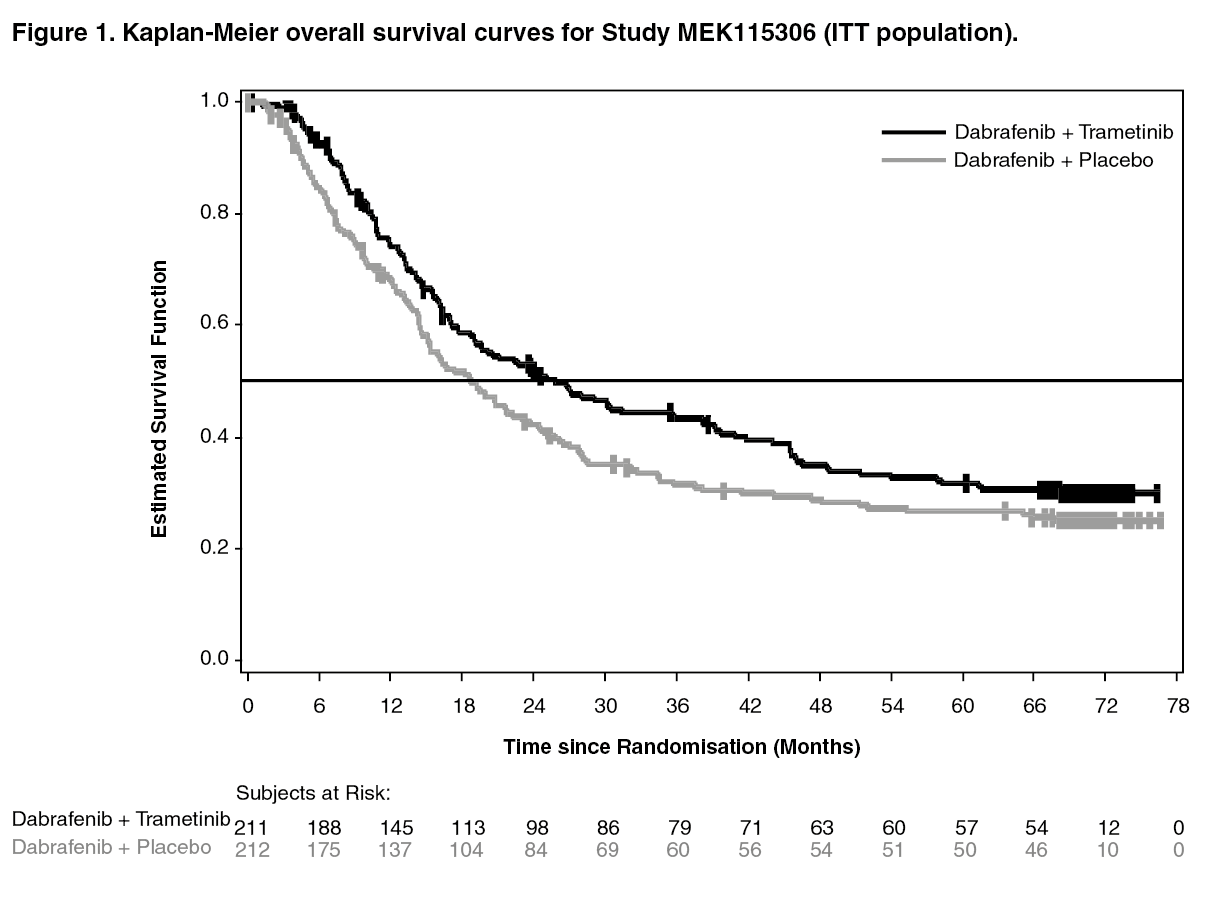 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image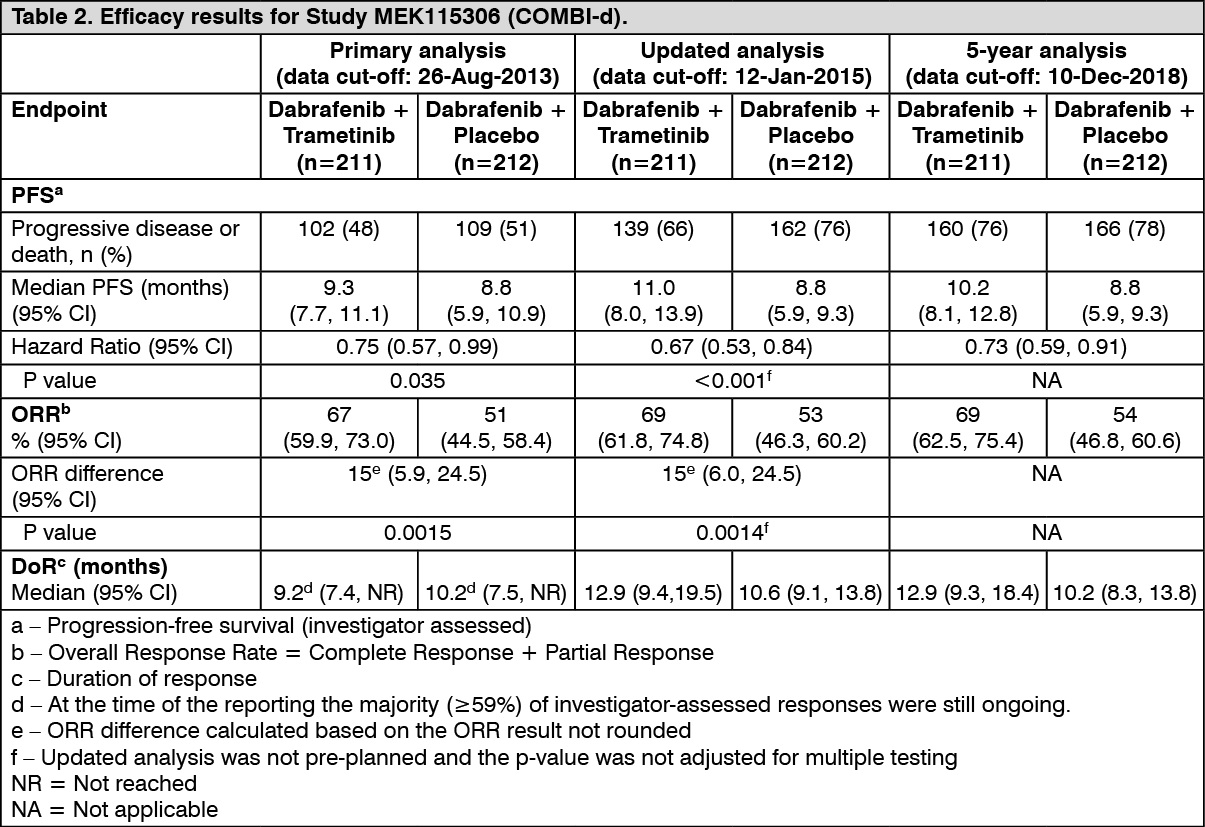 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image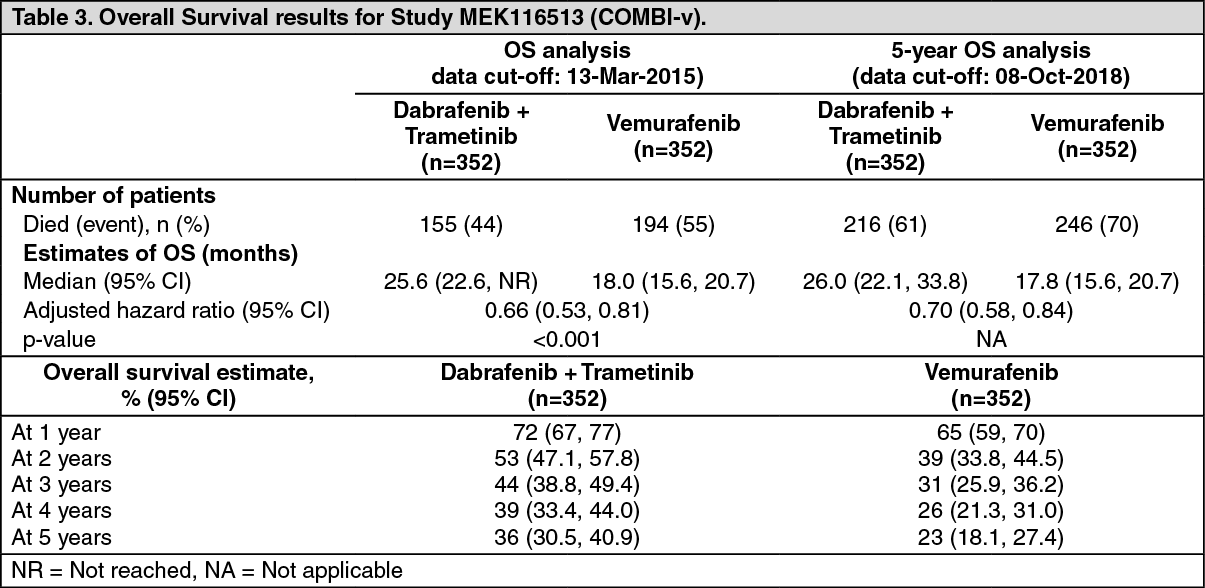 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image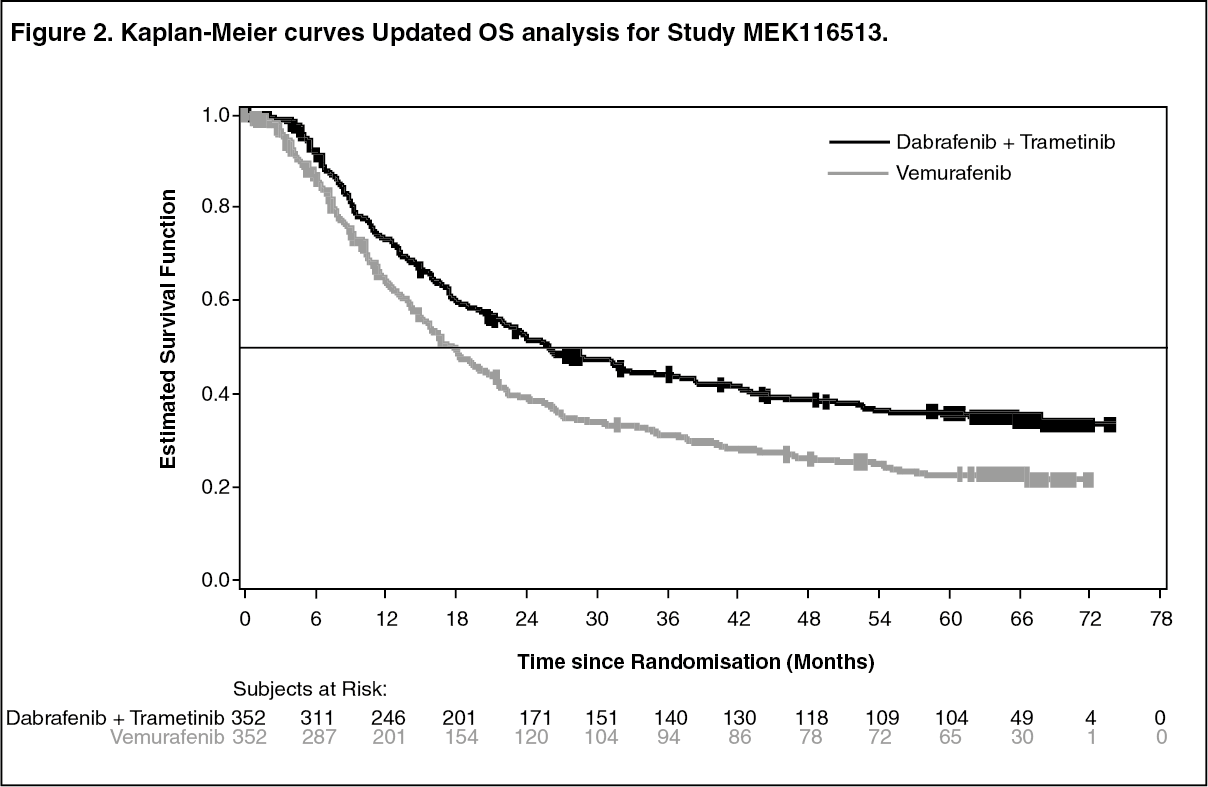 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image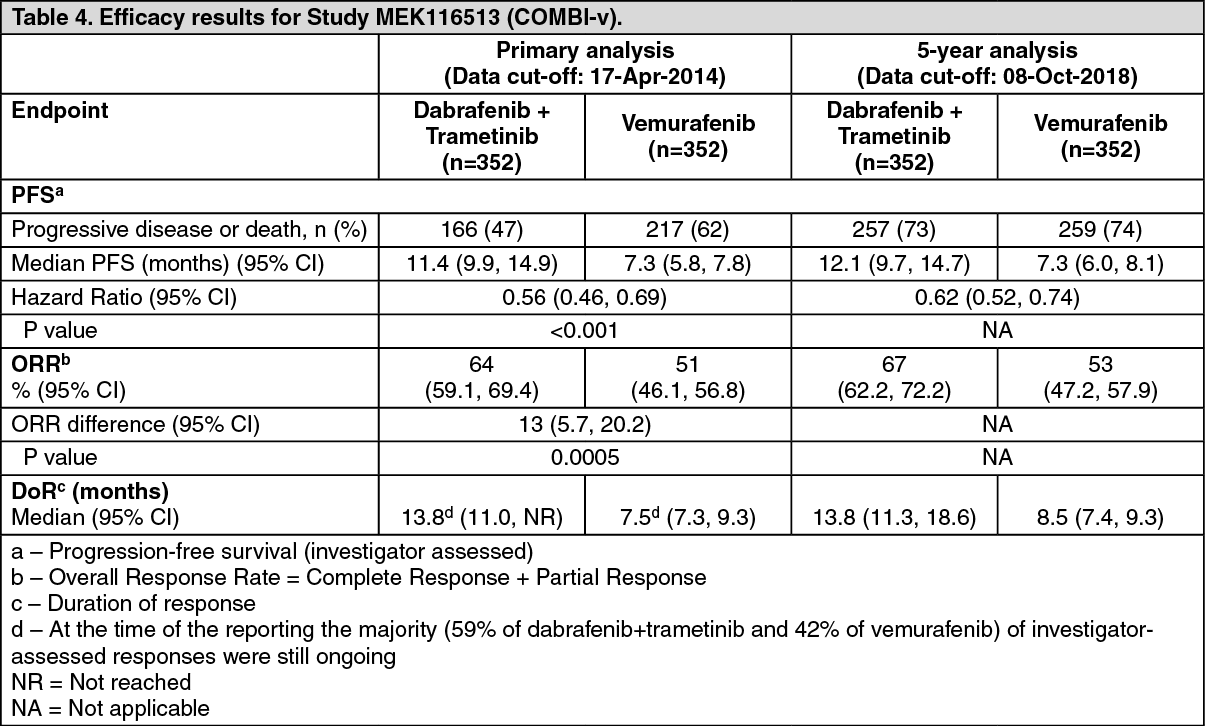 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image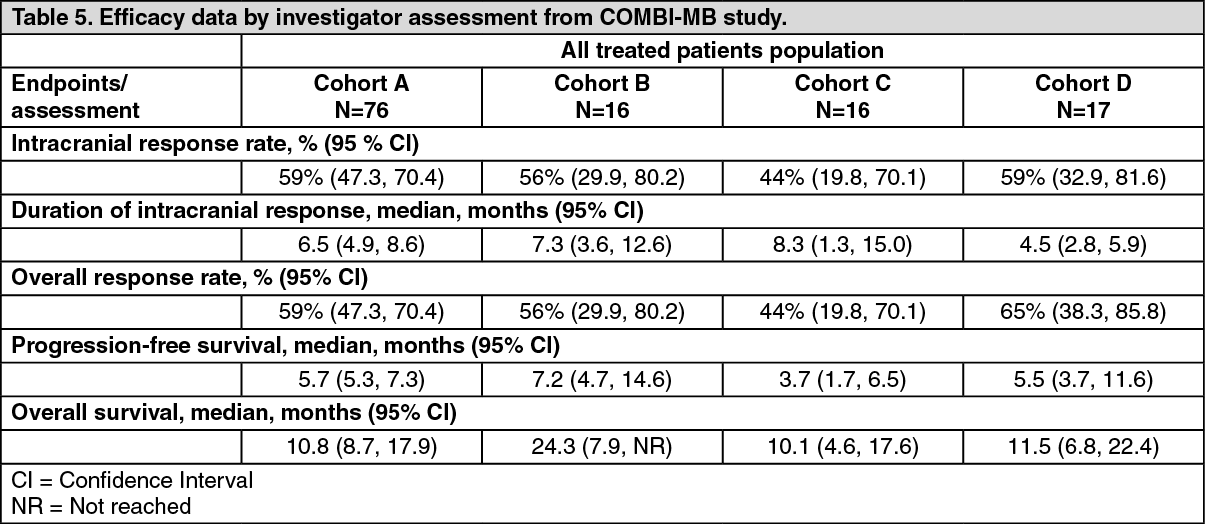 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image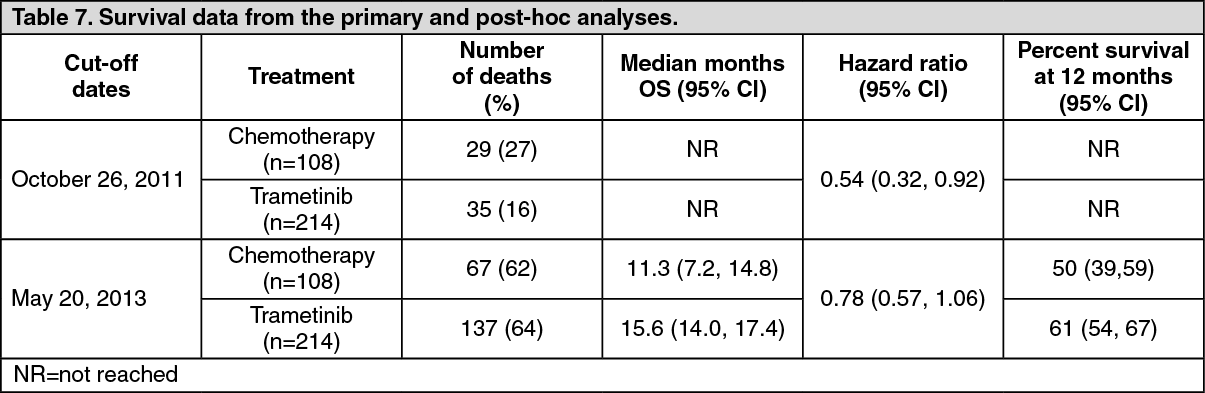 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image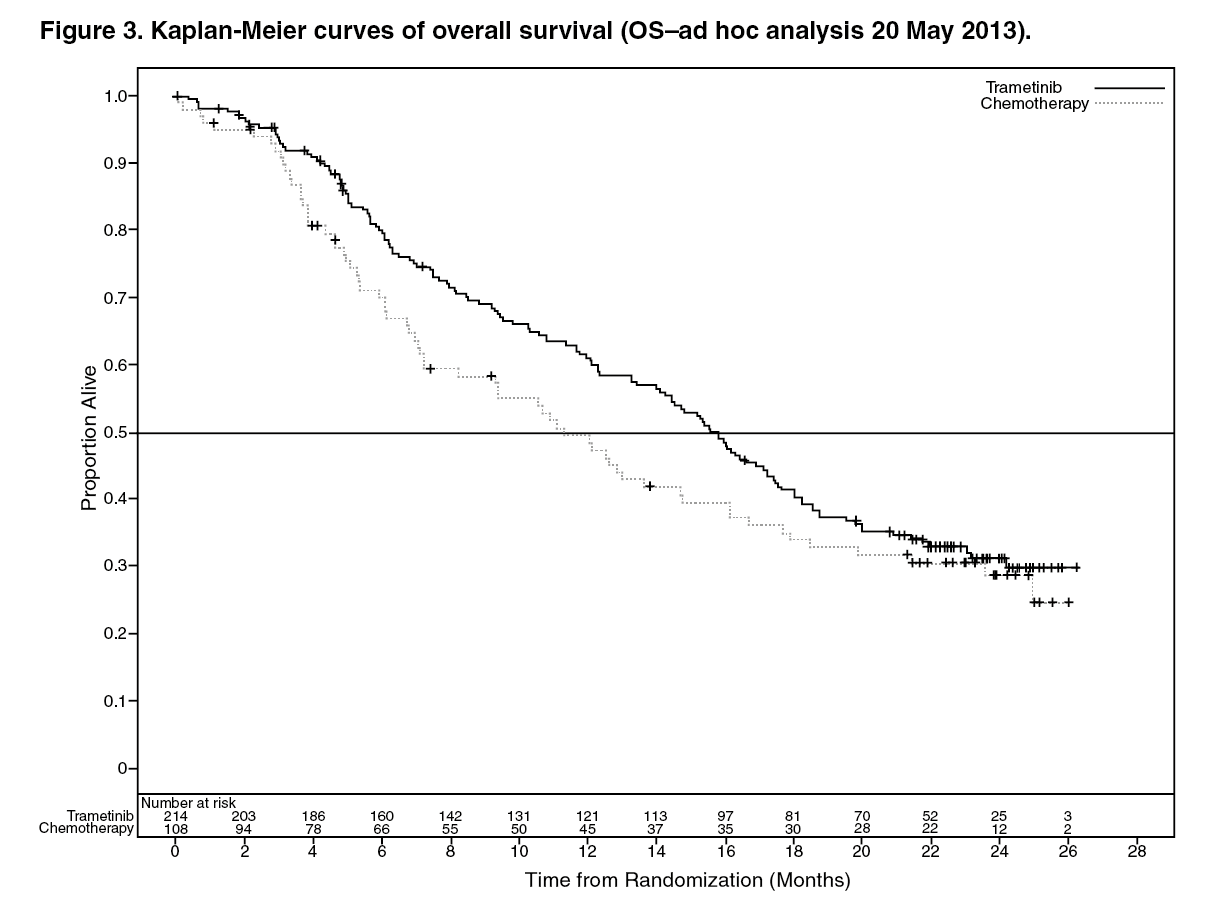 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image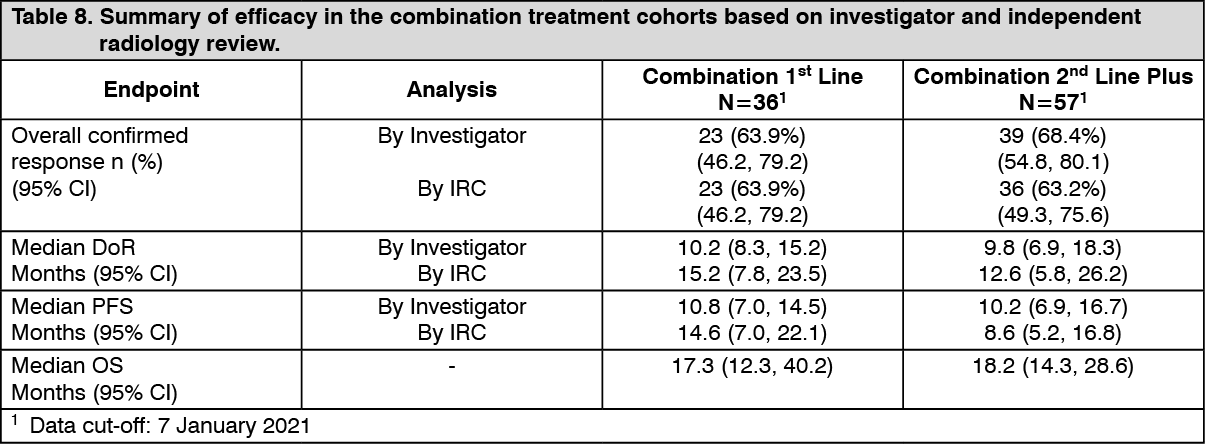 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image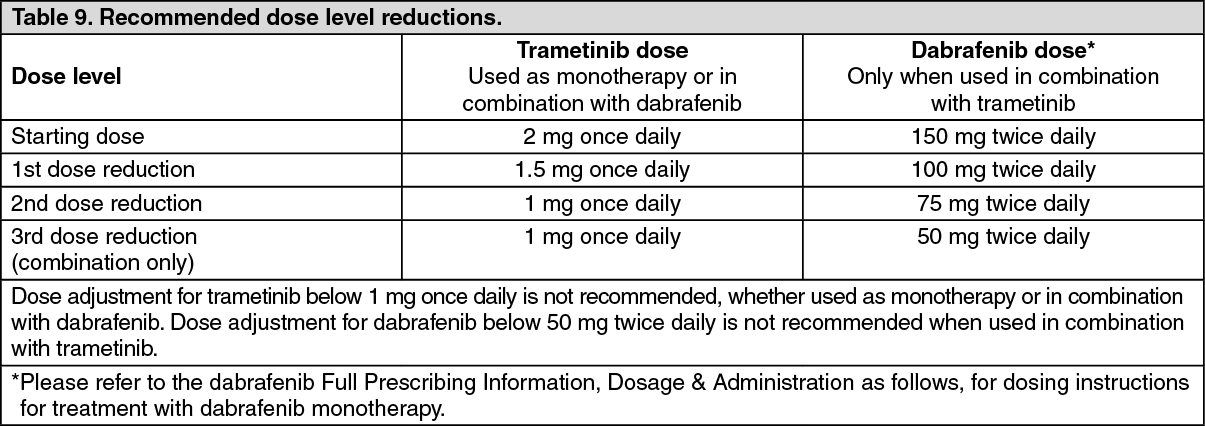 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image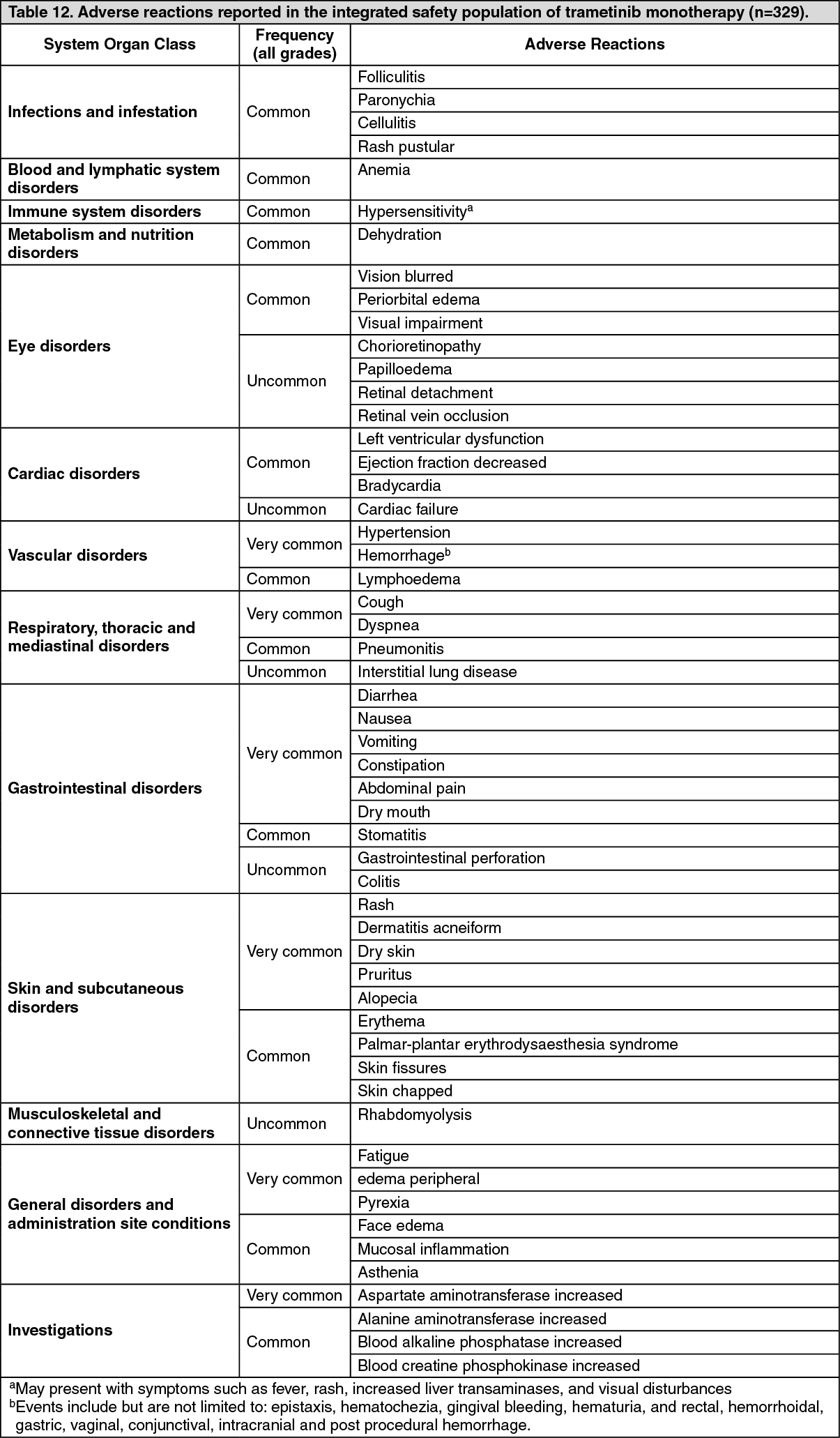 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image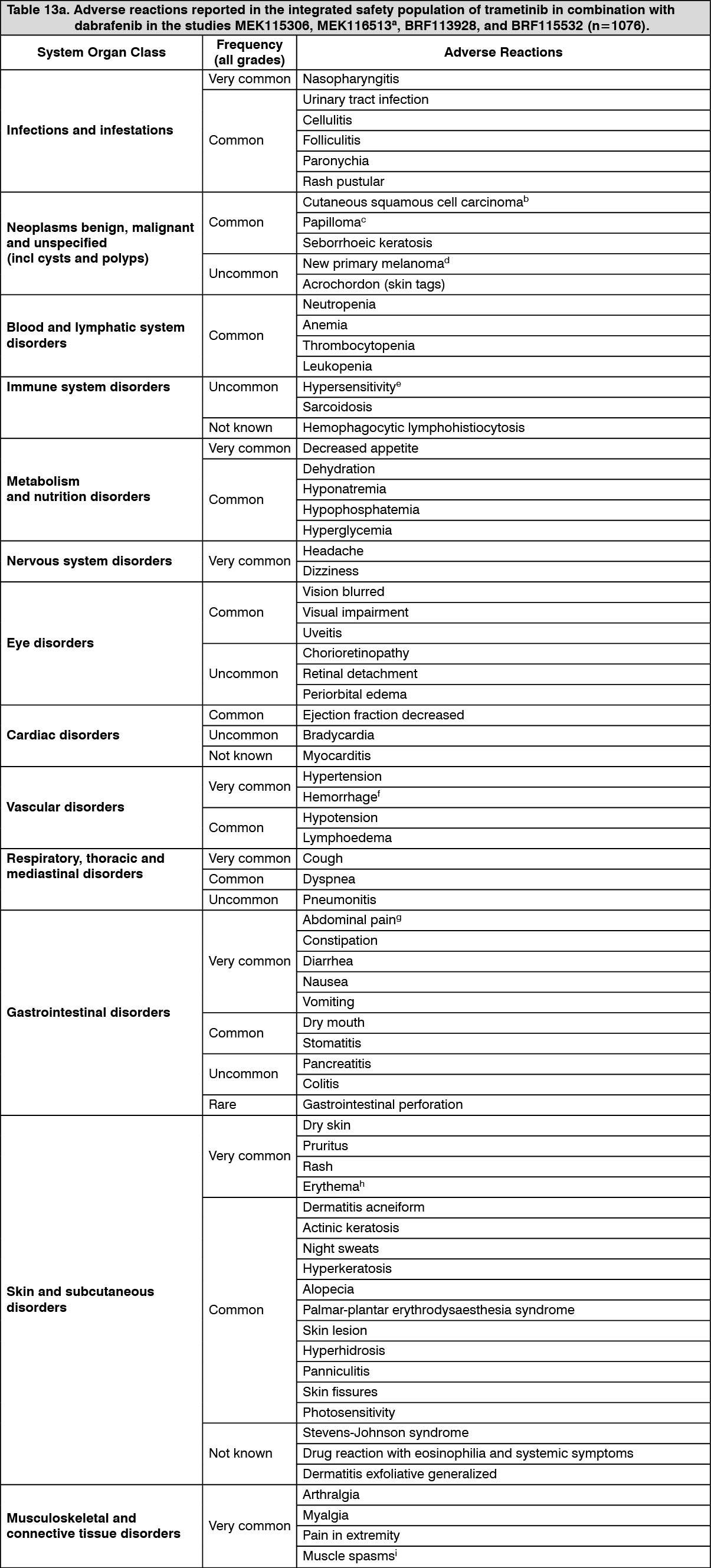 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image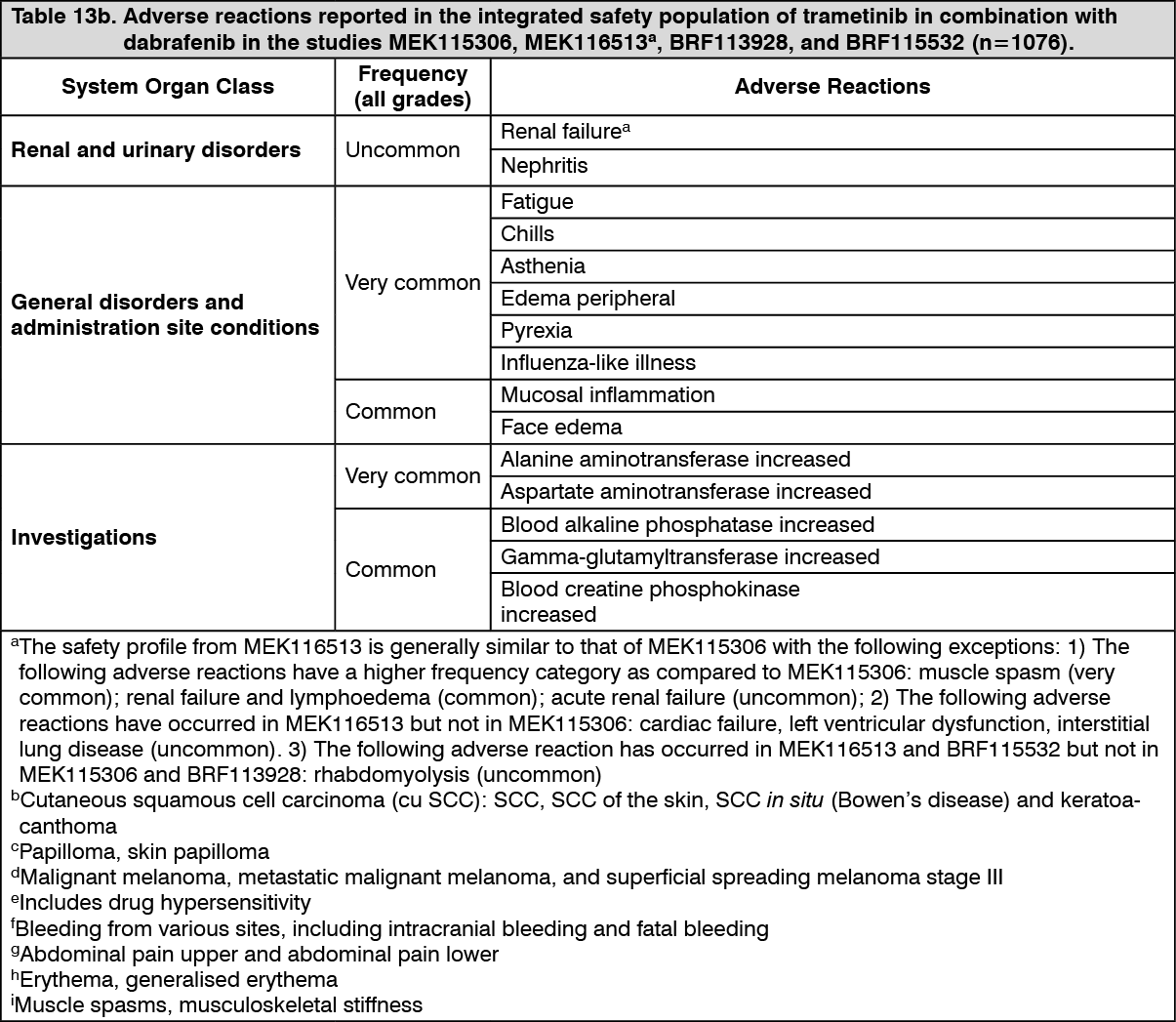 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out