


 Sign Out
Sign Out

Class I PI3K lipid kinases are key components of the PI3K/AKT/mTOR (mammalian target of rapamycin) signaling pathway.
Gain-of-function mutations in the gene encoding the catalytic alpha-subunit of PI3K (PIK3CA) lead to activation of PI3Kalpha manifested by increased lipid kinase activity, growth-factor independent activation of Akt-signaling, cellular transformation and the generation of tumors in a diverse array of preclinical models.
In vitro, alpelisib treatment potently inhibited the phosphorylation of PI3K downstream targets Akt as well as its various downstream effectors in breast cancer cells and showed selectivity towards cell lines harboring a PIK3CA mutation.
In vivo, alpelisib showed good tolerability as well as dose-and time-dependent inhibition of the PI3K/Akt pathway and dose-dependent tumor growth inhibition in relevant tumor xenograft models, including models of breast cancer.
PI3K inhibition by alpelisib treatment has been shown to induce an increase in ER transcription in breast cancer cells, therefore, sensitizing these cells to estrogen receptor (ER) inhibition by fulvestrant treatment. Combination of alpelisib and fulvestrant demonstrated increased anti-tumor activity than either treatment alone in xenograft models derived from ER+, PIK3CA mutated breast cancer cell lines (MCF-7 and KPL1).
Pharmacodynamics: In biochemical assays, alpelisib inhibited wild type PIK3alpha (IC50=4.6 nmol/L) and its 2 most common somatic mutations (H1047R, E545K) (IC50~4 nmol/L) more potently than the PI3Kdelta (IC50=290 nmol/L) and PI3Kgamma (IC50=250 nmol/L) isoforms and showed significantly reduced activity against PI3Kbeta (IC50=1,156 nmol/L).
The potency and selectivity of alpelisib was confirmed at the cellular level in mechanistic and relevant tumor cell lines.
Cardiac electrophysiology: Serial, triplicate ECGs (electrocardiogram) were collected following a single dose and at steady-state to evaluate the effect of alpelisib on the QTcF interval in patients with advanced cancer. A pharmacokinetic-pharmacodynamic analysis included a total of 134 patients treated with alpelisib at doses ranging from 30 to 450 mg.
The analysis demonstrates the absence of a clinically significant QTcF prolongation at the recommended 300 mg dose with or without fulvestrant. The estimated mean change from baseline in QTcF was <10 msecs (7.2 ms; 90% CI: 5.62, 8.83) at the observed geometric-mean Cmax at steady-state (2,900 ng/mL) following single agent administration at the recommended 300 mg dose.
Clinical Studies: Alpelisib (Pivikto) was evaluated in a pivotal phase III, randomized, double-blind, placebo controlled study of alpelisib (Pivikto) in combination with fulvestrant in men and postmenopausal women with HR+, HER2- locally advanced breast cancer whose disease had progressed or recurred on or after an aromatase inhibitor based treatment (with or without CDK4/6 combination).
A total of 572 patients were enrolled into two cohorts, cohort with PIK3CA mutation or cohort without PIK3CA mutation breast cancer. PIK3CA mutation status was determined by clinical trial assays. Patients were randomized to receive either alpelisib (Pivikto) 300 mg plus fulvestrant or placebo plus fulvestrant in a 1:1 ratio. Randomization was stratified by presence of lung and/or liver metastasis and previous treatment with CDK4/6 inhibitor(s).
Within the cohort with a PIK3CA mutation, 169 patients were randomized to receive alpelisib (Pivikto) in combination with fulvestrant and 172 patients were randomized to placebo in combination with fulvestrant. Within this cohort, 170 (49.9%) patients had liver/lung metastases and 20 (5.9%) patients had received prior CDK4/6 inhibitor treatment.
Within the cohort without PIK3CA mutation, 115 patients were randomized to receive alpelisib (Pivikto) in combination with fulvestrant and 116 were randomized to receive placebo in combination with fulvestrant. 112 (48.5%) patients had liver/lung metastases and 15 (6.5%) patients had prior CDK4/6 inhibitor treatment.
In the cohort with PIK3CA mutation, 97.7% of patients received prior hormonal therapy and 47.8% of patients had the last setting as metastatic and 51.9% of patients whose last setting was adjuvant therapy. Overall, 85.6% of the patients were considered to have endocrine resistant disease; primary endocrine resistance was observed in 13.2% and secondary endocrine resistance in 72.4% of patients.
In both cohorts with or without PIK3CA mutation, demographics and baseline disease characteristics, ECOG ( Eastern Cooperative Oncology Group) performance status, tumor burden, and prior antineoplastic therapy were well balanced between the study arms.
During the randomized treatment phase, alpelisib (Pivikto) 300 mg or alpelisib (Pivikto) matching placebo was administered orally once daily on a continuous basis. Fulvestrant 500 mg was administered intramuscularly on Cycle 1 Day 1 and 15 and then at Day 1 of a 28-day cycle during treatment phase (administration +/- 3 days).
Patients were not allowed to cross over from placebo to alpelisib (Pivikto) during the study or after disease progression.
The primary endpoint for the study was progression-free survival (PFS) using Response Evaluation Criteria in Solid Tumors (RECIST v1.1), based on the investigator assessment in patients with a PIK3CA mutation. The key secondary endpoint was overall survival (OS) for patients with a PIK3CA mutation.
Other secondary endpoints included PFS for patients without a PIK3CA mutation, OS for patients without a PIK3CA mutation, as well as overall response rate (ORR) and clinical benefit rate (CBR) by PIK3CA mutation cohort.
Cohort with PIK3CA mutation: Patients enrolled with a PIK3CA mutation had a median age of 63 years (range 25 to 92). 44.9% patients were 65 years of age or older and <85 years. The patients included were White (66.3%), Asian (21.7%), Black or African American (1.2%).
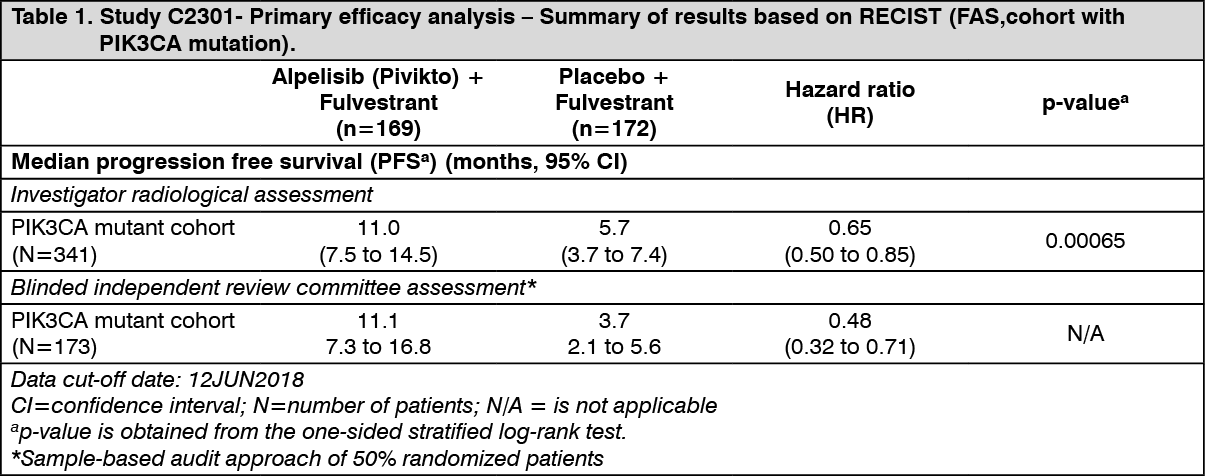
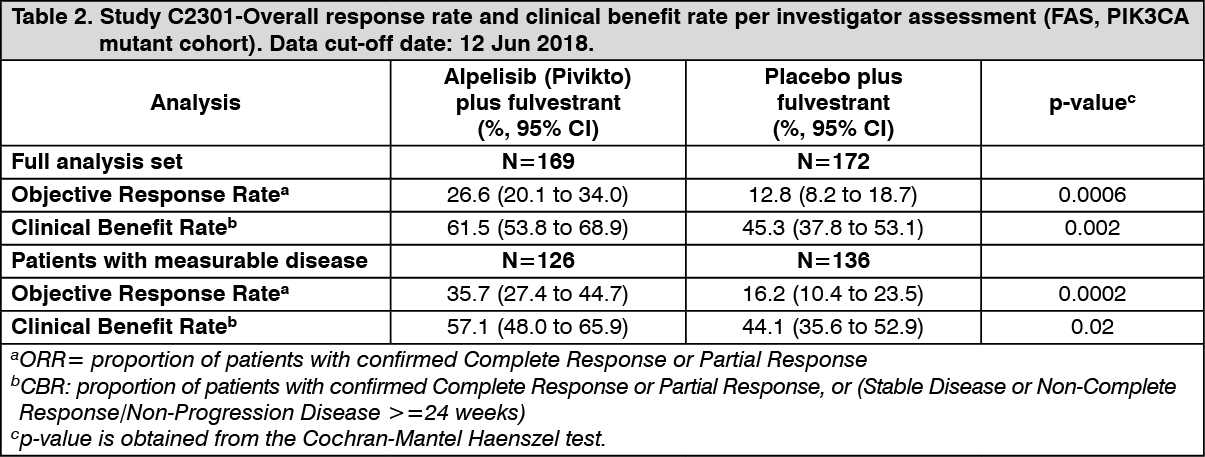
Primary analysis: The study met its primary objective at the final PFS analysis (data cut-off date 12-June-2018) demonstrating statistically significant improvement in PFS by investigator assessment in the PIK3CA mutant cohort for patients receiving alpelisib (Pivikto) plus fulvestrant, compared to patients receiving placebo plus fulvestrant (HR = 0.65 with 95% CI: 0.50, 0.85; one sided stratified log-rank test p= 0.00065), with an estimated 35% risk reduction of disease progression or death in favor of treatment with alpelisib (Pivikto) plus fulvestrant. The median PFS was prolonged by 5.3 months, from 5.7 months (95% CI: 3.7, 7.4) in the placebo plus fulvestrant arm to 11 months (95% CI: 7.5, 14.5) in the alpelisib (Pivikto) plus fulvestrant arm.
Primary PFS results were supported by consistent results from a blinded independent review committee (BIRC) assessment in this cohort, which included a randomly selected subset of 50% of randomized patients (HR= 0.48 with 95% CI: 0.32, 0.71).
PFS results are summarized in Table 1, Figure 1 and 2. (See Table 1 and Figures 1 and 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image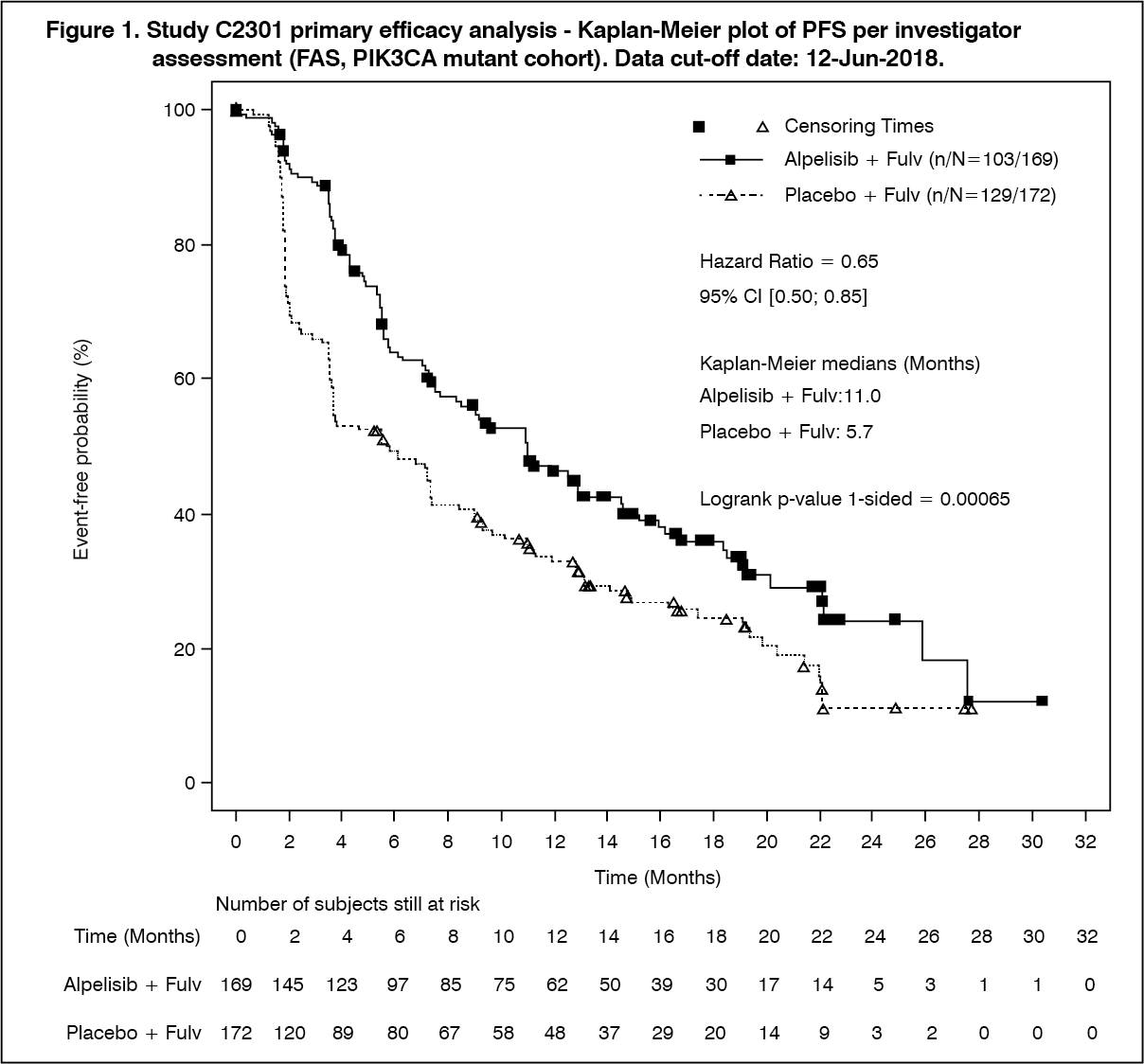 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image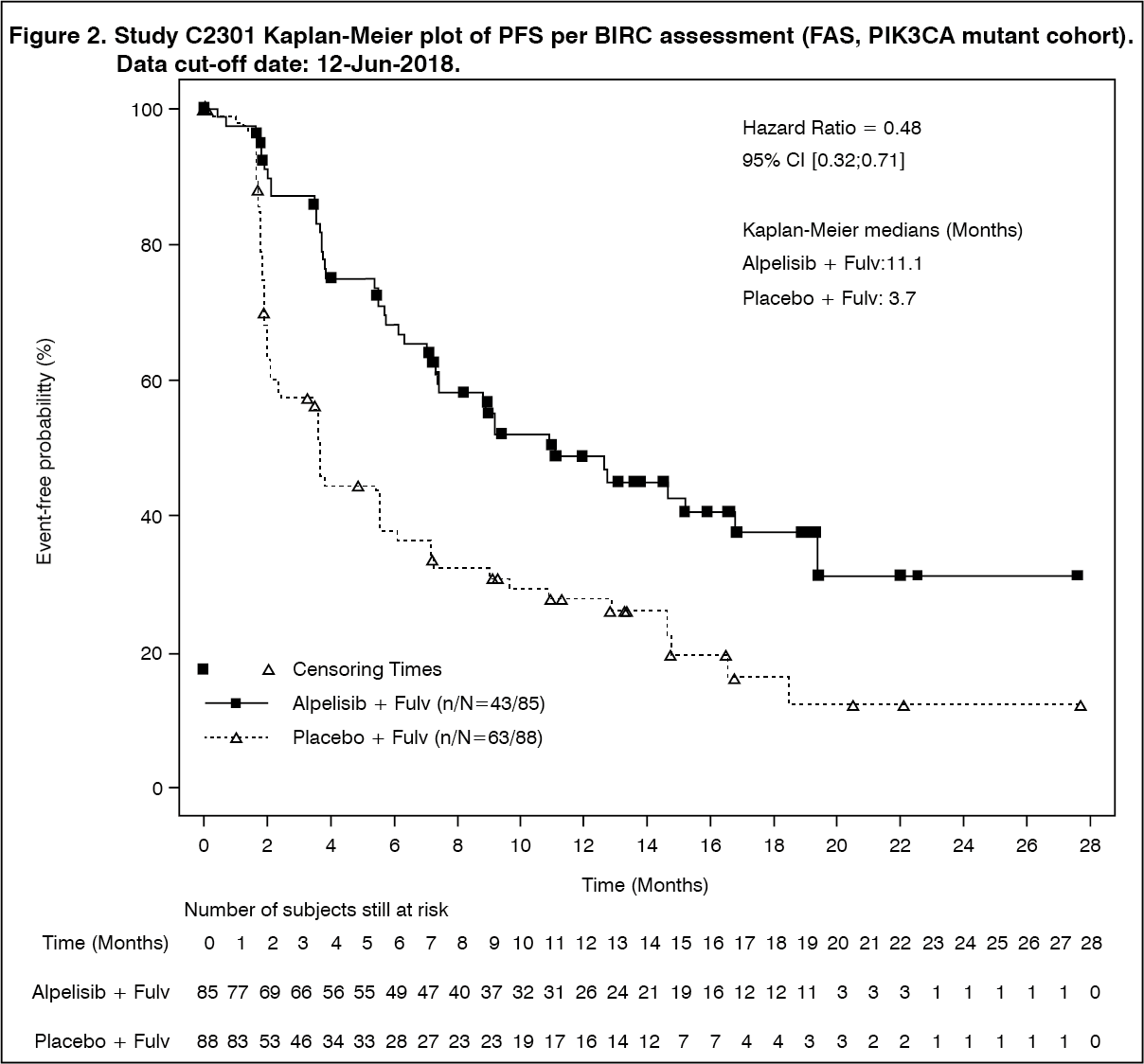 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePFS subgroup analyses by randomization stratification factors demonstrated a homogeneous and generally consistent treatment effect per investigator assessment across major demographic and prognostic subgroups irrespective of CDK4/6i prior treatment and presence or absence of lung/liver metastases.
Although limited in patient numbers, for the analysis of prior CDK4/6i treatment sub-group, the HR (95% CI) for PFS was 0.48 (0.17, 1.36).
In the subgroup of patients with presence of lung/liver metastases, the HR (95% CI) was 0.62 (0.44, 0.89). See Figure 3.
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageTreatment with the combination of alpelisib (Pivikto) plus fulvestrant was associated with marked improvements in ORR and CBR relative to placebo + fulvestrant. See Table 2 for details. (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe global health status/Quality of Life (QoL) outcomes were similar between the alpelisib (Pivikto) plus fulvestrant arm and the placebo plus fulvestrant arm. Time-to-Deterioration (TTD) in global health status EORTC QLQ-C30 (European Organization for Research and Treatment of Cancer Quality-of-life Questionnaire Core 30) was defined as time between baseline and first occurrence of ≥10 point worsening of global health status (EORTC QLQ-C30 global health scale score) compared to baseline with no later improvement above this threshold observed during the treatment period or death due to any cause. The addition of alpelisib (Pivikto) to fulvestrant showed no relevant difference in TTD in EORTC QLQ-C30 global health scale score compared with placebo plus fulvestrant, (HR=1.03; 95% CI:0.72, 1.48).
Final OS Analysis: The final OS analysis was conducted using a data cut-off date of 23-Apr-2020 and PFS was re-run using this data cut. With a median duration from randomization to data cut-off of approximately 42 months, the PFS benefit was sustained and consistent with results from the final PFS analysis. There was an estimated 36% risk reduction of progression or death in favor of treatment with alpelisib (Pivikto) plus fulvestrant (HR = 0.64; 95% CI: 0.50, 0.81). See Figure 4 for details. (See Figure 4.)
 Click on icon to see table/diagram/image
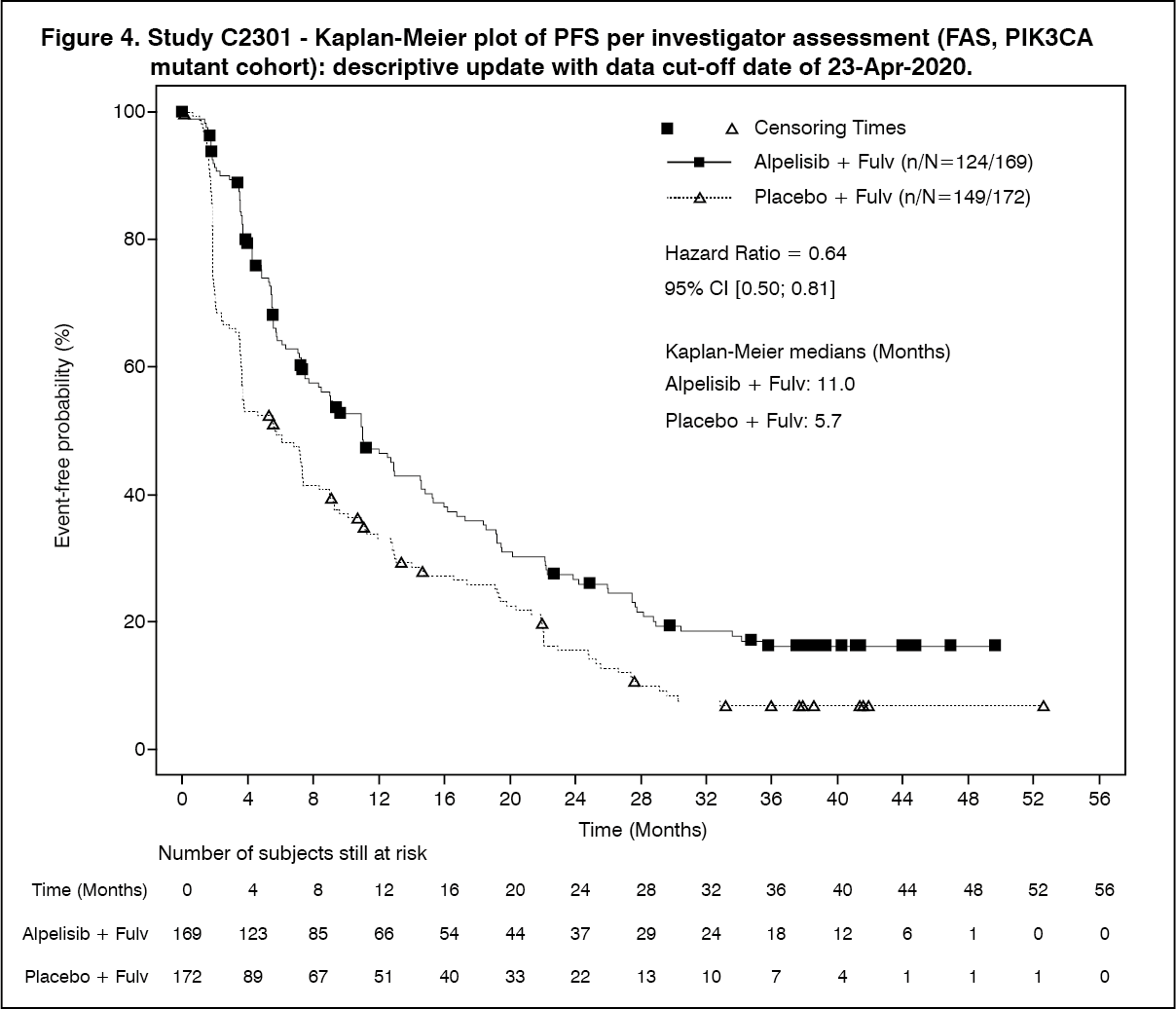
Click on icon to see table/diagram/imageAt the final OS analysis, the study did not meet its key secondary objective. As of the data cut-off date of 23-Apr-2020, a total of 87 (51.5%) deaths were reported in the alpelisib (Pivikto) plus fulvestrant arm and 94 (54.7%) in the placebo plus fulvestrant arm. The HR was 0.86 (95% CI: 0.64, 1.15; p = 0.15, one-sided) and the pre-specified O'Brien-Fleming efficacy boundary of p ≤ 0.0161 was not crossed. Median OS was prolonged by a clinically relevant 7.9 months, from 31.4 months (95% CI: 26.8, 41.3) in the placebo plus fulvestrant arm to 39.3 months (95% CI: 34.1, 44.9) in the alpelisib (Pivikto) plus fulvestrant arm. See Figure 5 for details. (See Figure 5.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageOS subgroup analyses by randomization stratification factors demonstrated a homogeneous and generally consistent treatment effect per investigator assessment. Although limited in patient numbers, for the analysis of prior CDK4/6i treatment subgroup, median OS in the alpelisib (Pivikto) plus fulvestrant arm was 29.8 months (95% CI: 6.7, 38.2) compared to 12.9 months (95% CI: 2.5, 34.6) in the placebo plus fulvestrant arm. In the subgroup of patients with presence of lung/liver metastases, median OS in the alpelisib (Pivikto) plus fulvestrant arm was 37.2 months (95% CI: 28.7, 43.6) compared to 22.8 months (95% CI: 19, 26.8) in the placebo plus fulvestrant arm.
Cohort without PIK3CA mutation: The proof of concept criteria to conclude a treatment benefit with alpelisib (Pivikto) and fulvestrant with respect to PFS in patients in the PIK3CA non-mutant cohort were not met (HR = 0.85; 95% CI: 0.58, 1.25) (see DOSAGE & ADMINISTRATION).
Pharmacokinetics: The pharmacokinetics of alpelisib were investigated in patients under an oral dosing regimen ranging from 30 to 450 mg daily. Healthy subjects received single oral doses ranging from 300 mg to 400 mg. The PK was mostly comparable in both oncology patients and healthy subjects.
Absorption: Following oral administration of alpelisib, median time to reach peak plasma concentration (Tmax) ranged between 2.0 to 4.0 hours, independent of dose, time or regimen. Based on absorption modelling bioavailability was estimated to be very high (>99%) under fed conditions but lower under fasted conditions (~68.7% at a 300 mg dose). Steady-state plasma levels of alpelisib after daily dosing can be expected to be reached on day 3, following onset of therapy in most patients.
Food effect: Alpelisib absorption is affected by food. In healthy volunteers after a single 300 mg oral dose of alpelisib, compared to the fasted state, a high-fat high-calorie (HFHC) meal (985 calories with 58.1 g of fat) increased AUCinf by 73% and Cmax by 84%, and a low-fat low-calorie (LFLC) meal (334 calories with 8.7 g of fat) increased AUCinf by 77% and Cmax by 145%. No significant difference was found for AUCinf between LFLC and HFHC with a geometric mean ratio of 0.978 [CI: 0.876, 1.09] showing that neither fat content nor overall caloric intake has a considerable impact on absorption. The increase in gastrointestinal solubility by bile, secreted in response to food intake, is considered to be the driver of the food effect. Hence, alpelisib (Pivikto) should be taken immediately after food, at approximately the same time each day.
Acid reducing agents: The co-administration of the H2 receptor antagonist ranitidine in combination with a single 300 mg oral dose of alpelisib slightly reduced the bioavailability of alpelisib and decreased overall exposure of alpelisib. In the presence of a LFLC meal, AUCinf was decreased on average by 21% and Cmax by 36% with ranitidine. In the absence of food, the effect was more pronounced with a 30% decrease in AUCinf and a 51% decrease in Cmax with ranitidine compared to the fasted state without co-administration of ranitidine. Alpelisib (Pivikto) can be co-administered with drugs that are acid-reducing agents, if alpelisib (Pivikto) is taken immediately after food. Population pharmacokinetic analysis showed no significant effect on the PK of alpelisib (Pivikto) by co-administration of acid reducing agents including proton pump inhibitors, H2 receptor antagonists and antacids.
Distribution: Alpelisib moderately binds to protein with a free fraction of 10.8% regardless of concentration. Alpelisib was equally distributed between red blood cells and plasma with a mean in vivo blood-to-plasma ratio of 1.03. There was no evidence for distribution into red blood cells caused by metabolites. Alpelisib did not penetrate the blood-brain-barrier in rats. As alpelisib (Pivikto) is a substrate of human efflux transporters, penetration of the blood-brain-barrier is not expected to occur in human. The volume of distribution of alpelisib at steady-state (Vss/F) is estimated at 114 L (intersubject CV% 46%).
Biotransformation/metabolism: In vitro studies demonstrated that formation of the hydrolysis metabolite BZG791 by chemical and enzymatic amide hydrolysis was a major metabolic pathway, followed by CYP3A4 mediated hydroxylation. Alpelisib hydrolysis occurs systemically by both chemical decomposition and enzymatic hydrolysis via ubiquitously expressed, high-capacity enzymes (esterases, amidases, choline esterase) not limited to the liver. CYP3A4-mediated metabolites and glucuronides amounted to ~15% of the dose BZG791 accounted for ~40 to 45% of the dose. The rest of the dose, which was found as unchanged alpelisib in urine and feces, was either excreted as alpelisib or non-absorbed.
Elimination: Alpelisib exhibits low clearance with 9.2 L/hr (CV% 21%) based on population PK analysis under fed conditions. The population derived half-life, independent of dose and time, was 8 to 9 hours at steady state of 300 mg, once daily.
In human mass-balance study, after oral administration, alpelisib and its metabolites were primarily found in the feces (81.0%), as alpelisib or metabolized as BZG791. Excretion in the urine is minor (13.5%), with 2% of unchanged alpelisib. Following single oral dose of [14C]- alpelisib, 94.5% of the total administered radioactive dose was recovered within 8 days.
Linearity/non-linearity: The pharmacokinetics were found to be linear with respect to dose and time under fed conditions between 30 and 450 mg. After multiple doses, alpelisib exposure (AUC) at steady-state is only slightly higher than that of a single dose with an average accumulation of 1.3 to 1.5 with a daily dosing regimen.
Metabolic interaction: Based on the results of metabolic in vitro induction and inhibition studies, alpelisib may induce the metabolic clearance of co-medications metabolized by CYP2B6, CYP2C9 and CYP3A4 and may inhibit the metabolic clearance of co-medications metabolized CYP3A4 (time-dependent inhibition) if sufficiently high concentrations are achieved in vivo.
In a drug-drug interaction study, co-administration of alpelisib with everolimus, a sensitive CYP3A4 substrate, confirmed that there are no clinically significant pharmacokinetic interactions (increase in AUC by 11.2%) between alpelisib and CYP3A4 substrates. No change in everolimus exposure was observed at alpelisib doses ranging from 250 to 300 mg, also confirmed by PBPK modeling with everolimus and midazolam (≤ 15% increase in AUC). Due to the concurrent induction and time-dependent inhibition by alpelisib, PBPK simulations with substrates of CYP3A4 that also possess an additional time-dependent inhibition and induction potential on CYP3A4 that affects their own metabolism predict changes in exposure (decrease or increase) less than 2-fold, depending on the substrate.
In a drug-drug interaction study, co-administration of alpelisib with rifampin, a strong CYP3A4 inducer, confirmed that there is a clinically significant pharmacokinetic interaction between alpelisib and strong CYP3A4 inducers leading to a decrease in AUC by 57% and 74% for a single 300 mg dose and a repeated 300 mg dose of alpelisib, respectively (see INTERACTIONS).
CYP2C9 substrates: In lieu of a clinical study, PBPK modeling showed that AUC and Cmax ratios of warfarin (10 mg single dose) were estimated to be 0.91 and 0.99, respectively, after repeated co-administration of alpelisib (300 mg), indicating no or weak induction potential of alpelisib on CYP2C9.
Transporter-based interaction: Alpelisib showed only weak in vitro inhibition towards the ubiquitously expressed efflux transporters (P-gp, BCRP, MRP2, BSEP), solute carrier transporters at the liver inlet (OATP1B1, OATP1B3, OCT1) and solute carrier transporters in the kidney (OAT1, OAT3, OCT2, MATE1, MATE2K. As unbound systemic steady state concentrations (or concentrations at the liver inlet) at both the therapeutic dose and maximum tolerated dose are significantly lower than the experimentally determined unbound inhibition constants or IC50, the inhibition will not translate into clinical significance. A clinically relevant effect on P-gp substrates can be excluded.
Fulvestrant: Data from a clinical study in patients with breast cancer indicated no effect of fulvestrant on alpelisib exposure (and vice versa) following co-administration of the drugs.
Special populations: Effect of age, weight and gender: The population PK analysis showed that there are no clinically relevant effects of age, body weight, or gender on the systemic exposure of alpelisib that would require alpelisib (Pivikto) dose adjustment.
Pediatric patients (below 18 years): The pharmacokinetics of alpelisib (Pivikto) in pediatric patients have not been established.
Geriatric patients (65 years or above): Of 284 patients who received alpelisib (Pivikto) in the phase III study (in alpelisib (Pivikto) plus fulvestrant arm), 117 patients were ≥65 years of age and 34 patients were ≥75 years of age. No overall differences in safety or effectiveness of alpelisib (Pivikto) were observed between these patients and younger patients (see DOSAGE & ADMINISTRATION).
Race/Ethnicity: Population PK analyses and PK analysis from a single agent study in Japanese cancer patients showed that there are no clinically relevant effects of ethnicity on the systemic exposure of alpelisib (Pivikto).
Non-compartmental PK parameters after single and multiple daily doses of alpelisib (Pivikto) for Japanese patients were very similar to those reported in the Caucasian population.
Renal impairment: No dose adjustment is necessary in patients with mild or moderate renal impairment. Patients with severe renal impairment have not been studied and caution should be used. Based on a population pharmacokinetic analysis that included 117 patients with normal renal function (eGFR [estimated glomerular filtration rate] ≥90 mL/min/1.73 m2) / (CLcr ≥90 mL/min), 108 patients with mild renal impairment (eGFR 60 to <90 mL/min/1.73m2)/ (CLcr 60 to <90 mL/min), and 45 patients with moderate renal impairment (eGFR 30 to <60 mL/min/1.73 m2), mild and moderate renal impairment had no effect on the exposure of alpelisib (see DOSAGE & ADMINISTRATION).
Hepatic impairment: No dose adjustment is necessary in patients with mild, moderate or severe hepatic impairment (Child-Pugh A, B and C).
Based on a pharmacokinetic trial in patients with hepatic impairment, moderate and severe hepatic impairment had negligible effect on the exposure of alpelisib (see DOSAGE & ADMINISTRATION). The mean exposure for alpelisib was increased by 1.26-fold in patients with severe (GMR [geometric mean ratio]: 1.00 for Cmax; 1.26 for AUClast/AUCinf) hepatic impairment.
Based on a population pharmacokinetic analysis that included 230 patients with normal hepatic function, 45 patients with mild hepatic impairment and no patients with moderate hepatic impairment, further supporting the findings from the dedicated hepatic impairment study, mild and moderate hepatic impairment had no effect on the exposure of alpelisib, (see DOSAGE & ADMINISTRATION).
Toxicology: Non-Clinical Safety Data: Alpelisib was evaluated in safety pharmacology, single- and repeated dose toxicity, genotoxicity and photo-toxicity studies.
Safety pharmacology and repeat dose toxicity: The majority of the observed alpelisib effects were related to the pharmacological activity of alpelisib as a p110alpha specific inhibitor of the PI3K pathway, such as the influence on the glucose homeostasis resulting in hyperglycemia and the risk of increased blood pressure.
The bone marrow and lymphoid tissue, pancreas, and some reproductive organs of both genders were the main target organs for adverse effects, which were generally reversible upon cessation of treatment. Alpelisib showed no effect on neuronal or pulmonary function. No evidence of skin irritation or corrosion was observed with alpelisib.
Cardiovascular safety pharmacology: In an in vitro hERG (human Ether-à-go-go-Related Gene) test, (where functionality of the human cardiac hERG channel heterologously expressed in HEK293 cells in vitro is assessed), an IC50 of 9.4 microM (4.2 microgram/mL) was found. No relevant electrophysiological effect was seen in dogs in several studies, up to single doses of 180 mg/kg in-vivo. An in vivo telemetry study in dogs showed an elevated blood pressure, starting at exposure lower than the exposure in humans, at the highest recommended dose of 300 mg/day.
Carcinogenicity and mutagenicity: No carcinogenicity studies have been conducted.
Alpelisib was not mutagenic in a Salmonella reverse mutation test in five strains, or aneugenic or clastogenic in human cell micronucleus and chromosome aberration tests in vitro. Also, an in vivo micronucleus test in peripheral blood reticulocytes obtained in week 4 of a 13-week rat repeated-dose toxicity study at dose levels of up to 20 mg/kg/day, at plasma exposure levels of about 1.7 times the exposure in humans at the highest recommended dose of 300 mg/day based on AUC, was negative.
Fertility and Reproductive toxicity: Please see USE IN PREGNANCY & LACTATION.
In repeated-dose toxicity studies up to 13 weeks duration, adverse effects were observed in reproductive organs of females and males, such as vaginal atrophy and oestrus cycle variations in rats (at or above 6 mg/kg/day, a dose that provides plasma exposure levels below the exposure in humans, at the highest recommended dose of 300 mg/day based on AUC), or prostate atrophy in dogs (at 15 mg/kg/day, at plasma exposure levels about 2.8 times the exposure in humans, at the highest recommended dose of 300 mg/day based on AUC) (see USE IN PREGNANCY & LACTATION). In general, the observed effects were reversible upon treatment discontinuation.
In fertility studies conducted in male and female rats, similar effects on fertility were observed. In females at doses of 20 mg/kg/day (approximately 1.7 times the estimated exposure (AUC) in humans at the recommended dose of 300 mg), increased pre- and post-implantation losses led to reduced numbers of implantation sites and live embryos. The NOAEL (No-observed-adverse- effect-level) for female fertility was determined at 10 mg/kg/day (at exposure levels (AUC) at or below the recommended human dose of 300 mg). In males, at doses of ≥ 10 mg/kg/day, accessory glands weights (seminal vesicles, prostate) were reduced and correlated microscopically with atrophy and/or reduced secretion in prostate and seminal vesicles, respectively. Male fertility parameters were unaffected at doses up to 20 mg/kg/day.
Toxicokinetic endpoints were not included in either of the two fertility studies since the toxicokinetic parameters of alpelisib in rats had been sufficiently established earlier.
Phototoxicity: An in vitro phototoxicity test on the on the mouse Balb/c 3T3 fibroblast cell line did not identify a relevant phototoxicity potential for alpelisib.
Juvenile animal studies: Juvenile animal studies are not available.