


 Sign Out
Sign Out
By increasing the endogenous levels of these incretin hormones, vildagliptin enhances the sensitivity of beta cells to glucose resulting in improved glucose-dependent insulin secretion. Treatment with 50 to 100 mg daily in patients with type 2 diabetes significantly improved markers of beta-cell function. The degree of improvement in beta-cell function is dependent on the initial degree of impairment; in non-diabetic (normal glycemic) individuals, vildagliptin does not stimulate insulin secretion or reduce glucose levels.
By increasing endogenous GLP-1 levels, vildagliptin enhances the sensitivity of alpha cells to glucose, resulting in more glucose-appropriate glucagon secretion. The reduction in inappropriate glucagon during meals in turn attenuates insulin resistance.
The enhanced increase in the insulin/glucagon ratio during hyperglycemia due to increased incretin hormone levels results in a decrease in fasting and postprandial hepatic glucose production, leading to reduced glycemia.
The known effect of increased GLP-1 levels to delay gastric emptying is not observed with vildagliptin treatment. In addition, a reduction in postprandial lipemia that is not associated with vildagliptin's incretin mediated effect to improve islet function, has been observed.
Mechanism of action: Vildagliptin, a member of the islet enhancer class, is a potent and selective dipeptidylpeptidase-4 (DPP-4) inhibitor that improves glycemic control. Vildagliptin inhibition of DPP-4 results in increased fasting and postprandial endogenous levels of the incretin hormones GLP-1 (glucagon-like peptide 1) and GIP (glucose-dependent insulinotropic polypeptide).
Clinical Studies: More than 15,000 patients with type 2 diabetes participated in double-blind, placebo or active-controlled clinical trials of up to more than 2 years of treatment duration. In these studies, vildagliptin was administered to more than 9,000 patients at daily doses of 50 mg once daily, 50 mg twice daily or 100 mg once daily. More than 5,000 male and more than 4,000 female patients received vildagliptin 50 mg once daily or 100 mg daily. More than 1,900 patients receiving vildagliptin 50 mg once daily or 100 mg daily were ≥65 years of age. In these trials, vildagliptin was administered as monotherapy in drug-naïve patients with type 2 diabetes or in combination in patients not adequately controlled by other antidiabetic medicinal products.
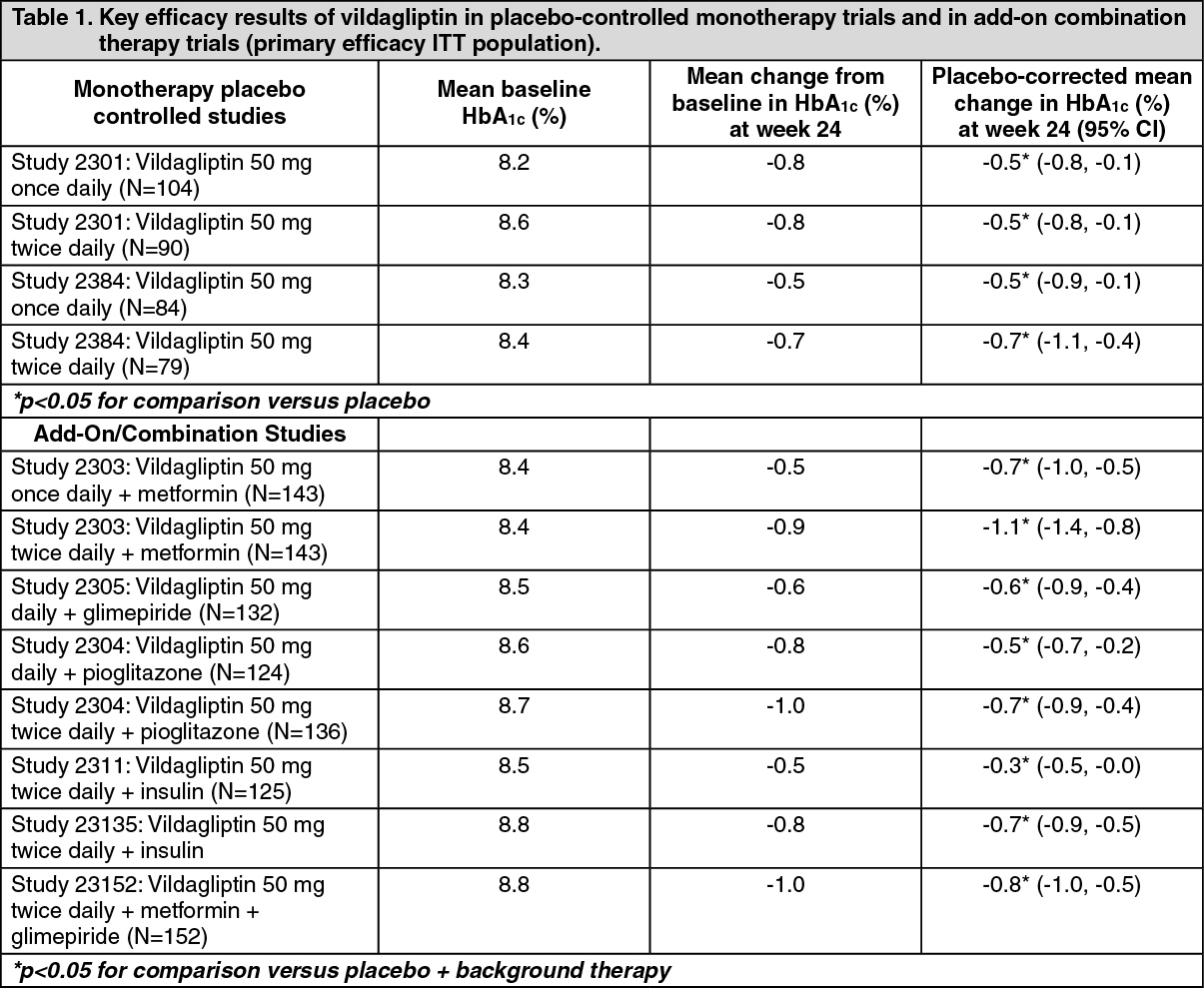
Overall, vildagliptin improved glycemic control when given as monotherapy or when used in combination with metformin, a sulfonylurea, a thiazolidinedione, with insulin, or in triple combination with metformin and a sulfonylurea as measured by clinically relevant reductions in HbA1c from baseline at the study endpoint (see Table 1).
In clinical trials, the magnitude of HbA1c reductions with vildagliptin was greater in patients with higher baseline HbA1c.
In a 52-week trial (LAF2309), vildagliptin (100 mg/day) reduced baseline HbA1c by ‑1% compared to -1.4% for metformin (titrated to 2 g/day). Patients treated with vildagliptin reported significantly lower incidences of gastrointestinal adverse reactions versus those treated with metformin.
In a 24-week trial (LAF2327), vildagliptin (100 mg/day) was compared to rosiglitazone (8 mg once daily). Mean reductions were -1.1% with vildagliptin and ‑1.3% with rosiglitazone in patients with mean baseline HbA1c of 8.7%. Patients receiving rosiglitazone experienced a mean increase in weight (+1.6 kg) while those receiving vildagliptin experienced no weight gain (‑0.3 kg). The incidence of peripheral edema was lower in the vildagliptin group than in the rosiglitazone group (2.1% vs. 4.1%, respectively).
In a 24-week trial (LAF2354) vildagliptin (50 mg twice daily) was compared to pioglitazone (30 mg once daily) in patients inadequately controlled with metformin.
Mean reductions from baseline HbA1c of 8.4% were -0.9% with vildagliptin added to metformin and -1.0% with pioglitazone added to metformin. The decrease in HbA1c from baseline >9.0% was greater (-1.5%) in both treatment groups. Patients receiving pioglitazone in addition to metformin experienced an increase in weight of 1.9 kg.
Patients receiving vildagliptin in addition to metformin experienced an increase in weight of 0.3 kg. In a 28-week extension, HbA1c reductions were similar between treatment groups and the difference in body weight further increased.
In a long-term trial of up to more than 2 years (LAF2308), vildagliptin (100 mg/day) was compared to glimepiride (up to 6 mg/day) in patients treated with metformin.
After one year, mean reductions in HbA1c were -0.4% with vildagliptin added to metformin and -0.5% with glimepiride added to metformin. Body weight change with vildagliptin was -0.2 kg vs +1.6 kg with glimepiride. The incidence of hypoglycemia was significantly lower in the vildagliptin group (1.7%) than in the glimepiride group (16.2%). At study endpoint (2 years), the HbA1c was similar to baseline values in both treatment groups and the body weight changes and the differences in hypoglycemia were maintained.
In a long-term trial of 2 years (LAF2310), vildagliptin (50 mg twice daily) was compared to gliclazide (up to 320 mg/day). After two years, mean reduction in HbA1c was -0.5% for vildagliptin and -0.6% for gliclazide. Vildagliptin had less of a weight gain (0.75 kg) and fewer hypoglycemic events (0.7%) than gliclazide (1.6 kg and 1.7%, respectively).
In a 52-week trial (LAF237A2338), vildagliptin (50 mg twice daily) was compared to gliclazide (up to 320 mg/day) in patients inadequately controlled with metformin. After one year, mean reductions in HbA1c were -0.81% with vildagliptin added to metformin (mean baseline HbA1c 8.4%) and -0.85% with gliclazide added to metformin (mean baseline HbA1c 8.5%); statistical non-inferiority was achieved. Body weight change with vildagliptin was +0.1 kg compared to a weight gain of +1.4 kg with gliclazide. The number of patients experiencing hypoglycemic events was the same in both treatment groups, however the number of patients experiencing two or more hypoglycemic events was higher in the gliclazide plus metformin group (0.8%) than in the vildagliptin plus metformin group (0.2%).
In a 24-week trial (LMF237A2302) the efficacy of the fixed dose combination of vildagliptin and metformin (gradually titrated to a dose of 50 mg/500 mg twice daily or 50 mg/1,000 mg twice daily) as initial therapy in drug-naive patients was evaluated.
The mean HbA1c reductions were significantly greater with vildagliptin plus metformin combination therapy compared to either monotherapy. Vildagliptin/metformin 50 mg/1,000 mg twice daily reduced HbA1c by -1.82% and vildagliptin/metformin 50 mg/500 mg twice daily by -1.61% from a mean baseline HbA1c of 8.6%. The decrease in HbA1c observed in patients with a baseline ≥10.0% was greater. Body weight decreased in all groups, with a mean reduction of -1.2 kg for both vildagliptin plus metformin combinations. The incidence of hypoglycemia was similar across treatment groups (0% with vildagliptin plus metformin combinations and 0.7% with each monotherapy).
In a 24-week double-blind placebo-controlled trial, vildagliptin (50 mg once daily) reduced HbA1c by -0.74% from a mean baseline of 7.9% in patients with moderate renal impairment and -0.88% from a mean baseline of 7.7% in patients with severe renal impairment. Vildagliptin significantly decreased HbA1c when compared to placebo (reductions in patients with moderate and severe renal impairment in the placebo group were -0.21% and -0.32% respectively, from similar mean baseline values).
A 24-week randomized, double-blind, placebo-controlled trial was conducted in 449 patients to evaluate the efficacy and safety of vildagliptin (50 mg twice daily) in combination with a stable dose of basal or premixed insulin (mean daily dose 41 U), with (N=276) or without (N=173) concomitant metformin.
Vildagliptin in combination with insulin significantly decreased HbA1c compared with placebo: In the overall population, the placebo-adjusted mean reduction from mean baseline HbA1c 8.8% was -0.72%. In the subgroups treated with insulin with or without concomitant metformin, the placebo-adjusted mean reduction in HbA1c was -0.63% and -0.84%, respectively. The incidence of hypoglycemia in the overall population was 8.4% and 7.2% in the vildagliptin and placebo groups, respectively. Changes in weight were +0.2 kg and -0.7 kg in the vildagliptin and placebo groups, respectively.
A 24-week randomized, double-blind, placebo-controlled study was conducted in 318 patients to evaluate the efficacy and safety of vildagliptin (50 mg twice daily) in combination with metformin (≥1,500 mg daily) and glimepiride (≥4 mg daily).
Vildagliptin in combination with metformin and glimepiride significantly decreased HbA1c compared with placebo: the placebo-adjusted mean reduction from mean baseline HbA1c 8.8% was -0.76%. (See Table 1).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageA 52-week multi-center, randomized, double-blind trial was conducted in patients with type 2 diabetes and congestive heart failure (CHF) (NYHA class I-III) to evaluate the effect of vildagliptin 50 mg bid (N=128) compared to placebo (N=126) on left ventricular ejection fraction (LVEF). Vildagliptin was not associated with a change in left-ventricular function or worsening of pre-existing CHF. Adjudicated cardiovascular events were overall balanced. There were slightly more cardiac events in vildagliptin treated patients with NYHA class III heart failure compared to placebo. However, there were imbalances in baseline CV risk favoring placebo and the number of events was low, precluding firm conclusions. Vildagliptin significantly decreased HbA1c compared with placebo (difference of 0.6%) from a mean baseline of 7.8%. The incidence of hypoglycemia in the overall population was 4.7% and 5.6% in the vildagliptin and placebo groups, respectively.
Cardiovascular risk: A meta-analysis of independently and prospectively adjudicated cardiovascular events from 37 phase III and IV monotherapy and combination therapy clinical studies of up to more than 2 years in duration was performed. It involved 9,599 patients with type 2 diabetes treated with vildagliptin 50 mg qd or 50 mg bid and showed that vildagliptin treatment was not associated with an increase in cardiovascular risk. The composite endpoint of adjudicated major adverse cardiovascular events (MACE) including acute myocardial infarction, stroke or CV death was similar for vildagliptin versus combined active and placebo comparators [Mantel-Haenszel risk ratio (M-H RR) 0.82 (95% confidence interval 0.61-1.11)] supporting the cardiovascular safety of vildagliptin. A MACE occurred in 83 out of 9,599 (0.86%) vildagliptin-treated patients and in 85 out of 7,102 (1.20%) comparator-treated patients. Assessment of each individual MACE component showed no increased risk (similar M-H RR). Confirmed HF events defined as HF requiring hospitalization or new onset of HF were reported in 41 (0.43%) vildagliptin-treated patients and 32 (0.45%) comparator-treated patients, with M-H RR 1.08 (95% CI 0.68-1.70) showing no increased risk of HF in vildagliptin-treated patients.
Pharmacokinetics: Absorption: Following oral administration in the fasting state, vildagliptin is rapidly absorbed with peak plasma concentrations observed at 1.75 hours. Co-administration with food slightly decreases the rate of absorption of vildagliptin, as characterized by a 19% decrease in peak concentrations, and a delay in the time to peak plasma concentration to 2.5 hours. There is no change in the extent of absorption, and food does not alter the overall exposure (AUC).
Distribution: The plasma protein binding of vildagliptin is low (9.3%), and vildagliptin is distributed equally between plasma and red blood cells. The mean volume of distribution of vildagliptin at steady state after intravenous administration (Vss) is 71 liters, suggesting extravascular distribution.
Biotransformation/Metabolism: Metabolism is the major elimination pathway for vildagliptin in humans, accounting for 69% of the dose. The major metabolite, LAY151, is pharmacologically inactive and is the hydrolysis product of the cyano moiety, accounting for 57% of the dose, followed by the amide hydrolysis product (4% of the dose). DPP-4 contributes partially to the hydrolysis of vildagliptin as shown in an in-vivo study using DPP-4 deficient rats.
Vildagliptin is not metabolized by cytochrome P450 enzymes to any quantifiable extent. In-vitro studies demonstrated that vildagliptin does not inhibit or induce cytochrome P450 enzymes.
Elimination: Following oral administration of [14C]- vildagliptin, approximately 85% of the dose is excreted into the urine and 15% of the dose is recovered in the feces. Renal excretion of unchanged vildagliptin accounts for 23% of the dose after oral administration.
After intravenous administration to healthy subjects, the total plasma and renal clearances of vildagliptin are 41 liters/hour and 13 liters/hour, respectively. The mean elimination half-life after intravenous administration is approximately 2 hours. The elimination half-life after oral administration is approximately 3 hours and is independent of the dose.
Linearity/Non-linearity: Vildagliptin is rapidly absorbed with an absolute oral bioavailability of 85%. Peak plasma concentrations for vildagliptin and the area under the plasma concentration versus time curve (AUC) increased in an approximately dose-proportional manner over the therapeutic dose range.
Special populations: Gender: No differences in the pharmacokinetics of vildagliptin were observed between male and female subjects with a diverse range of age and body mass index (BMI). DPP-4 inhibition by vildagliptin was unaffected by gender.
Obesity: BMI does not show any impact on the pharmacokinetic parameters of vildagliptin. DPP-4 inhibition by vildagliptin was unaffected by BMI.
Hepatic impairment: The effect of impaired hepatic function on the pharmacokinetics of vildagliptin was studied in subjects with mild, moderate, and severe hepatic impairment based on the Child-Pugh scores (ranging from 6 for mild to 12 for severe) in comparison to subjects with normal hepatic function. The exposure to vildagliptin (100 mg) after a single dose in subjects with mild and moderate hepatic impairment was decreased (20% and 8%, respectively), while the exposure to vildagliptin for subjects with severe impairment was increased by 22%. The maximum change (increase or decrease) in the exposure to vildagliptin is ~30%, which is not considered to be clinically relevant.
There was no correlation between the severity of hepatic function impairment and changes in exposure to vildagliptin.
The use of vildagliptin is not recommended in patients with hepatic impairment including patients with a pre-treatment ALT or AST >2.5x ULN.
Renal impairment: The AUC of vildagliptin increased on average 1.4, 1.7 and 2-fold in patients with mild, moderate and severe renal impairment, respectively, compared to normal healthy subjects. The AUC of the metabolites LAY151 increased 1.6, 3.2 and 7.3-fold and that of BQS867 increased 1.4, 2.7 and 7.3-fold in patients with mild, moderate and severe renal impairment, respectively, compared to healthy volunteers. Limited data from patients with end stage renal disease (ESRD) indicate that vildagliptin exposure is similar to that in patients with severe renal impairment. LAY151 concentrations in ESRD patients were approximately 2 to 3-fold higher than in patients with severe renal impairment. Dosage adjustment may be required in patients with renal impairment. (see Dosage & Administration).
Vildagliptin was removed by hemodialysis to a limited extent (3% over a 3 to 4-hour hemodialysis session starting 4 hours post dose).
Geriatric patients (65 years or above): In otherwise healthy elderly subjects (≥70 years), the overall exposure to vildagliptin (100 mg once daily) was increased by 32% with an 18% increase in peak plasma concentration compared to younger healthy subjects (18 to 40 years). These changes are not considered to be clinically relevant. DPP-4 inhibition by vildagliptin is not affected by age in the age groups studied.
Pediatric patients (below 18 years): No pharmacokinetic data available.
Ethnicity: There was no evidence that ethnicity affects the pharmacokinetics of vildagliptin.
Toxicology: Non-Clinical Safety Data: Carcinogenicity and mutagenicity: A two-year carcinogenicity study was conducted in rats at oral doses up to 900 mg/kg (approximately 200 times the human exposure at the maximum recommended dose).
No increases in tumor incidence attributable to vildagliptin were observed. A two-year carcinogenicity study was conducted in mice at oral doses up to 1,000 mg/kg (up to 240 times the human exposure at the maximum recommended dose). Mammary tumor incidence was increased in female mice at approximately 150 times the maximum anticipated human exposure to vildagliptin; it was not increased at approximately 60 times the maximum human exposure. The incidence of hemangiosarcoma was increased in male mice treated at 42 to 240 times the maximum human exposure to vildagliptin and in female mice at 150 times the maximum human exposure. No significant increases in hemangiosarcoma incidences were observed at approximately 16 times the maximum human exposure to vildagliptin in males and approximately 60 times the maximum human exposure in females.
Vildagliptin was not mutagenic in a number of mutagenicity tests including a bacterial reverse mutation Ames assay and a human lymphocyte chromosomal aberration assay. Oral bone marrow micronucleus tests in both rats and mice did not reveal clastogenic or aneugenic potential up to 2,000 mg/kg or approximately 400 times the maximum human exposure. An in-vivo mouse liver comet assay using the same dose was also negative.
Safety pharmacology and repeat dose toxicity: In a 13-week toxicology study in cynomolgus monkeys, skin lesions have been recorded at doses ≥5 mg/kg/day. These were consistently located on the extremities (hands, feet, ears and tail). At 5 mg/kg/day (approximately equivalent to human AUC exposure at the 100 mg dose), only blisters were observed. They were reversible despite continued treatment and were not associated with histopathological abnormalities. Flaking skin, peeling skin, scabs and tail sores with correlating histopathological changes were noted at doses ≥20 mg/kg/day (approximately 3 times human AUC exposure at the 100 mg dose). Necrotic lesions of the tail were observed at ≥80 mg/kg/day. It should be noted that vildagliptin exhibits a significantly higher pharmacological potency in monkeys compared with humans. Skin lesions were not reversible in the monkeys treated at 160 mg/kg/day during a 4-week recovery period. Skin lesions have not been observed in other animal species or in humans treated with vildagliptin.