


 Sign Out
Sign Out

Pharmacology: Pharmacodynamics: Mechanism of action: Esketamine, the S‑enantiomer of racemic ketamine, is an antidepressant with a novel mechanism of action. It is a non-selective, non‑competitive antagonist of the N-methyl-D-aspartate (NMDA) receptor, an ionotropic glutamate receptor.
Putative etiological contributors of depression, including stress and other conditions, are known to cause structural and functional impairment of synapses in brain regions involved with the regulation of mood and emotional behavior. Evidence within the literature suggests that through NMDA receptor antagonism, esketamine produces a transient increase in glutamate release leading to increases in α‑amino‑3‑hydroxy‑5‑methyl‑4‑isoxazolepropionic acid receptor (AMPAR) stimulation and subsequently to increases in neurotrophic signaling that restore synaptic function in these brain regions. Unlike other antidepressant therapies, esketamine's primary antidepressant action does not directly involve monoamine, GABA, or opioid receptors.
Pharmacodynamic effects: Effect on driving: Two studies were conducted to assess the effects of Esketamine (Spravato) on driving skills, one study in adult subjects with major depressive disorder and one study in healthy subjects. On‑road driving performance was assessed by the mean standard deviation of the lateral position (SDLP), a measure of driving impairment.
A single‑blind, placebo‑controlled study in 25 adult subjects with major depressive disorder evaluated the effects of a single 84‑mg dose of esketamine nasal spray on next day driving. An ethanol‑containing beverage was used as a positive control. The SDLP after administration of single 84‑mg dose of esketamine nasal spray was similar to placebo. The upper limit of the two‑sided 95% confidence interval (CI) of the mean difference between single‑dose of esketamine and placebo was 0.58 cm, which is less than the pre‑specified non‑inferiority margin of 2.4 cm. The lower limit of the 95% CI of the mean difference between ethanol and placebo was 1.03 cm (p<0.001), verifying assay sensitivity.
A randomized, double‑blind, cross‑over, placebo‑controlled study in 23 healthy subjects evaluated the effects of a single 84‑mg dose of esketamine nasal spray on driving. Mirtazapine was used as a positive control. Driving performance was assessed at 8 hours after esketamine or mirtazapine administration. The SDLP after esketamine nasal spray administration was similar to placebo. The upper limit of the two‑sided 95% CI of the mean difference between esketamine and placebo was 0.86 cm, which is less than the pre‑specified non‑inferiority margin of 2.4 cm. The lower limit of the 95% CI of the mean difference between mirtazapine and placebo was 1.12 cm (p=0.001), verifying assay sensitivity. Of the 23 subjects evaluated, 21 subjects completed the test successfully. Two subjects discontinued the driving test after receiving esketamine because of a perceived inability to drive.
Effect on QT/QTc interval and cardiac electrophysiology: Treatment with Esketamine (Spravato) did not prolong the QTc interval. The effect of Esketamine (Spravato) (84 mg nasal spray and 0.8 mg/kg esketamine intravenously infused over 40 minutes) on the QTc interval was evaluated in a randomized, double‑blind, placebo‑, and positive‑controlled (moxifloxacin 400 mg), 4‑period, crossover study in 60 healthy subjects. Maximum esketamine concentrations in plasma produced by the intravenous infusion were approximately 3‑times higher than the maximum concentrations produced by the nasal dose of 84 mg. The upper bound of the 90% confidence interval for the largest placebo‑adjusted, baseline‑corrected QTc interval remained below 10 msec, at all evaluated time‑points, based on Fridericia's correction method (QTcF) for both treatment groups.
Clinical studies: The efficacy and safety of Esketamine (Spravato) nasal spray was evaluated in five Phase 3 clinical studies in adult patients (18 to 86 years) with treatment‑resistant depression (TRD) who met DSM‑5 criteria for major depressive disorder and were non‑responders to at least two oral antidepressants (ADs) treatments, of adequate dosage and duration, in the current major depressive episode. 1833 adult patients were enrolled, of which 1601 patients were exposed to Esketamine (Spravato).
Treatment‑resistant depression - Short‑term studies: Esketamine (Spravato) was evaluated in three Phase 3 short‑term (4‑week) randomized, double‑blind, multicenter, active‑controlled studies in patients with TRD. Studies TRANSFORM‑1 (TRD3001) and TRANSFORM‑2 (TRD3002) were conducted in adults (18 to <65 years) and Study TRANSFORM‑3 (TRD3005) was conducted in adults ≥65 years of age. Patients in TRD3001 and TRD3002 initiated treatment with Esketamine (Spravato) 56 mg plus a newly initiated daily oral AD or a newly initiated daily oral AD plus placebo nasal spray on Day 1 and Esketamine (Spravato) dosages were then maintained on 56 mg or titrated to 84 mg administered twice‑weekly during a 4‑week double‑blind induction phase. Esketamine (Spravato) doses of 56 mg or 84 mg were fixed in Study TRD3001 and flexible in Study TRD3002. In Study TRD3005, patients (≥65 years) initiated treatment with Esketamine (Spravato) 28 mg plus a newly initiated daily oral AD or a newly initiated daily oral AD plus placebo nasal spray (Day 1) which was maintained or titrated to 56 mg or 84 mg dose administered twice‑weekly during a 4‑week double‑blind induction phase. A newly initiated open‑label oral AD (SNRI: duloxetine, venlafaxine extended release; SSRI: escitalopram, sertraline) was initiated on Day 1 in all studies. The selection of the newly initiated oral AD was determined by the investigator based on the patient's prior treatment history.
The baseline demographic and disease characteristics of patients in TRD3001 and TRD3002 studies were similar between the Esketamine (Spravato) plus oral AD and oral AD plus placebo nasal spray groups. The median subject age was 47 years (range 18 to 64 years), 67% were female; 83% Caucasian and 5% of African descent and mean duration of prior AD treatment was approximately 425 days. At the time of screening, the mean duration of the current episode of depression was 168 weeks. At the time of screening, 90% of patients had non‑response to ≥2 oral ADs with the remainder requiring confirmation of non‑response to the second AD during the 4‑week screening prospective phase. The new open‑label oral AD initiated during the 4‑week double‑blind induction phase was an SSRI in 38% of patients and an SNRI in 62% of patients. In TRD3005, the median subject age was 69 years (range 65 to 86 years) of which, 85% of patients were 65‑74 years of age, 62% were female and 95% were Caucasian and mean duration of prior AD treatment was approximately 727 days. At the time of screening, the mean duration of the current episode of depression was 216 weeks in TRD3005. At the time of screening, 85% of patients had non‑response to ≥2 oral ADs with the remainder requiring confirmation of non‑response to the second AD during the 4‑week screening prospective phase. The new open‑label AD initiated during the 4‑week double‑blind induction phase was an SSRI in 55% of patients and an SNRI in 45% of patients.
The primary efficacy measure was change from baseline in the Montgomery‑Åsberg Depression Rating Scale (MADRS) total score at the end of the 4‑week double‑blind induction phase. The MADRS is a ten‑item, clinician‑rated scale used to assess severity of depressive symptoms. Scores on the MADRS range from 0 to 60, with higher scores indicating more severe depression.
In the flexible dose study TRD3002, for the primary efficacy measure of improvement in depressive symptoms (change in MADRS total scores from baseline at the end of the 4‑week induction phase), Esketamine (Spravato) plus a newly initiated oral AD demonstrated clinically meaningful and statistical superiority compared to standard of care (newly initiated oral AD) plus placebo nasal spray. In studies TRD3001 and TRD3005, a clinically meaningful treatment effect in change in MADRS total scores from baseline at the end of the 4‑week induction phase was observed favoring Esketamine (Spravato) plus newly initiated oral AD compared with standard of care (newly initiated oral AD) plus placebo nasal spray (Table 1 [MMRM]). In TRD3002, improvements in the Sheehan Disability Scale (SDS) total score assessing global functional impairment and Patient Health Questionnaire‑9 (PHQ‑9) total score assessing symptoms of depression numerically favored Esketamine (Spravato) plus a newly initiated oral AD compared to standard of care (newly initiated oral AD) plus placebo nasal spray. (See Table 1.)
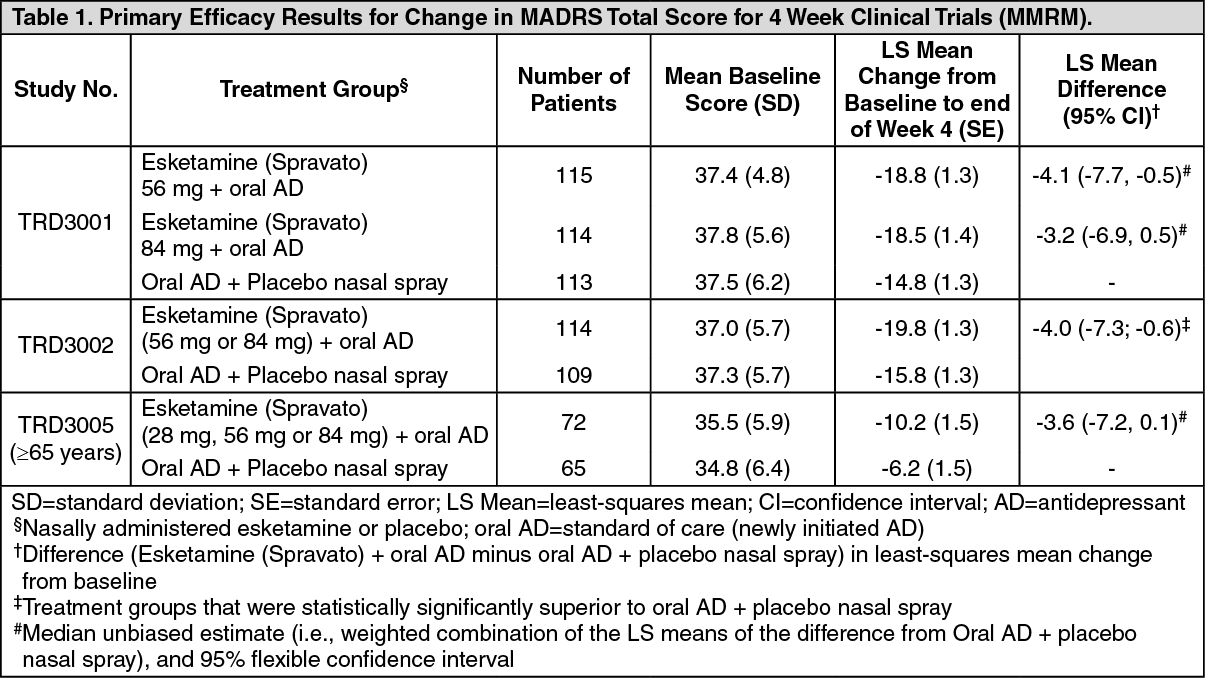 Click on icon to see table/diagram/image
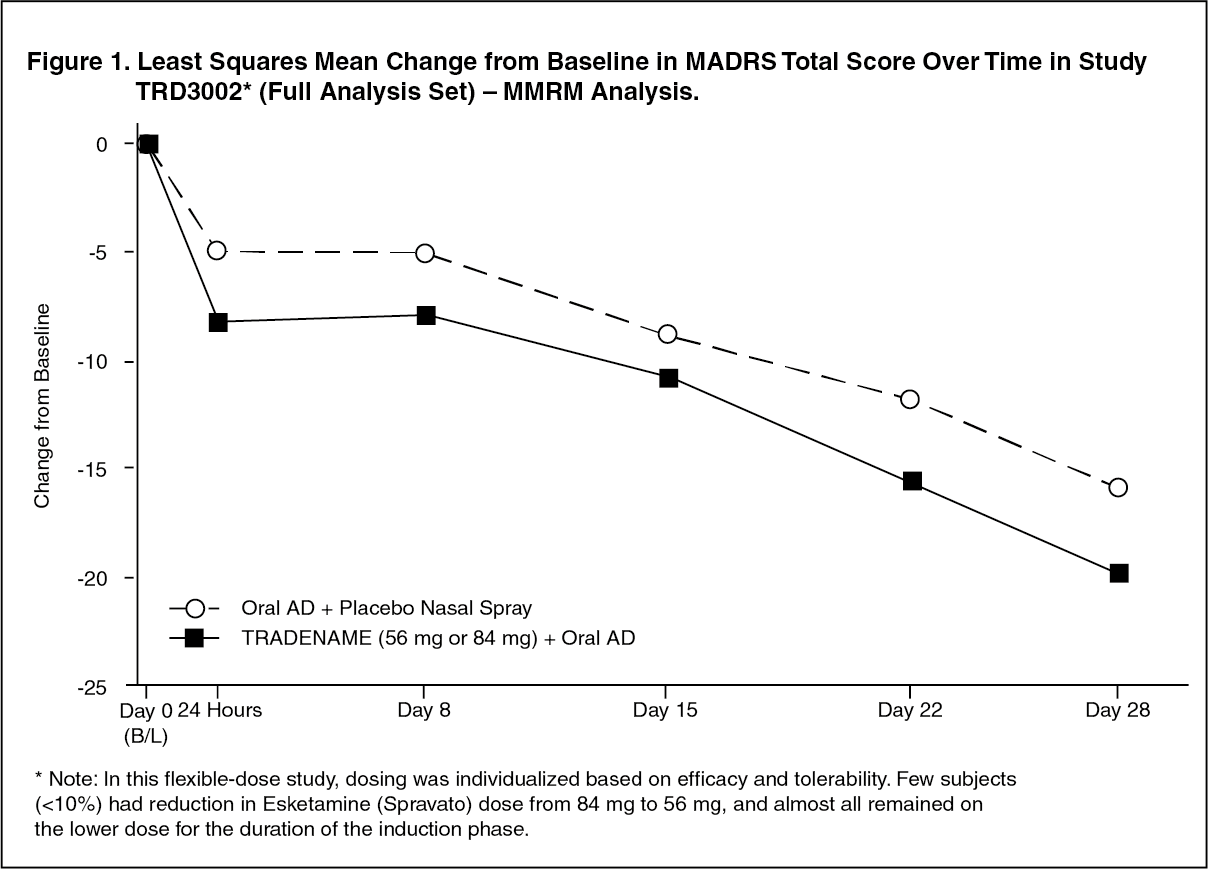
Click on icon to see table/diagram/imageTime Course of Treatment Response: In Study TRD3002, an effect of Esketamine (Spravato) on symptom reduction was observed as early as 24 hours post‑dose and increased in subsequent weeks with the full antidepressant effect of Esketamine (Spravato) seen by Day 28. Throughout the 4‑week double blind induction phase of Study TRD3002, the mean change in MADRS total score for flexibly dosed Esketamine (Spravato) (56 mg or 84 mg) plus oral AD was greater than for oral AD plus nasally‑administered placebo. At Day 28, 67% of the patients randomized to Esketamine (Spravato) were on 84 mg. Figure 1 [MMRM Analysis] depicts time course of response in the primary efficacy measure (MADRS) in Study TRD3002. A consistent treatment effect was observed in Studies TRD3001 and TRD3005. (See Figure 1.)
 Click on icon to see table/diagram/image
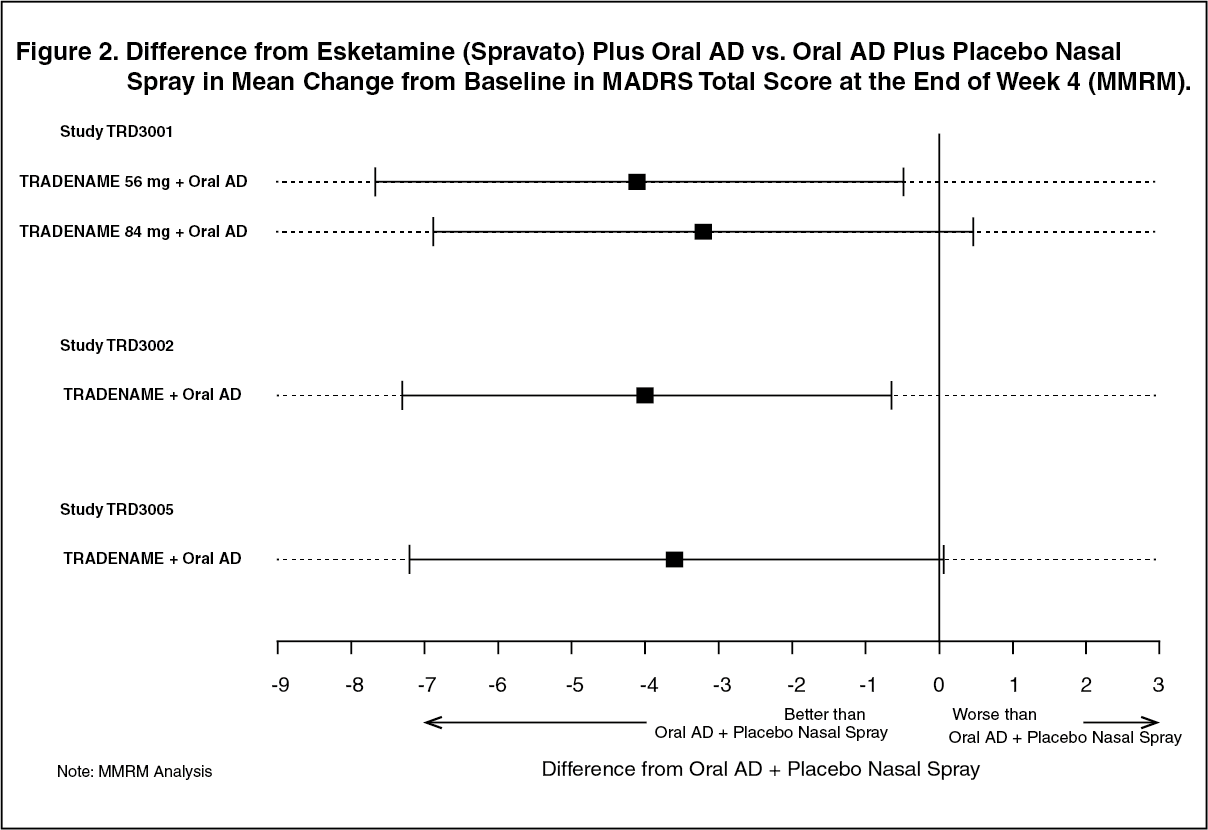
Click on icon to see table/diagram/imageIn addition, in TRD3002 study Esketamine (Spravato) plus oral AD was superior to oral AD plus placebo nasal spray in terms of mean change from baseline in MADRS total score at the end of Week 4. A consistent treatment difference was observed in Studies TRD3001 and TRD3005. (Figure 2 [MMRM]). (See Figure 2.)
 Click on icon to see table/diagram/image
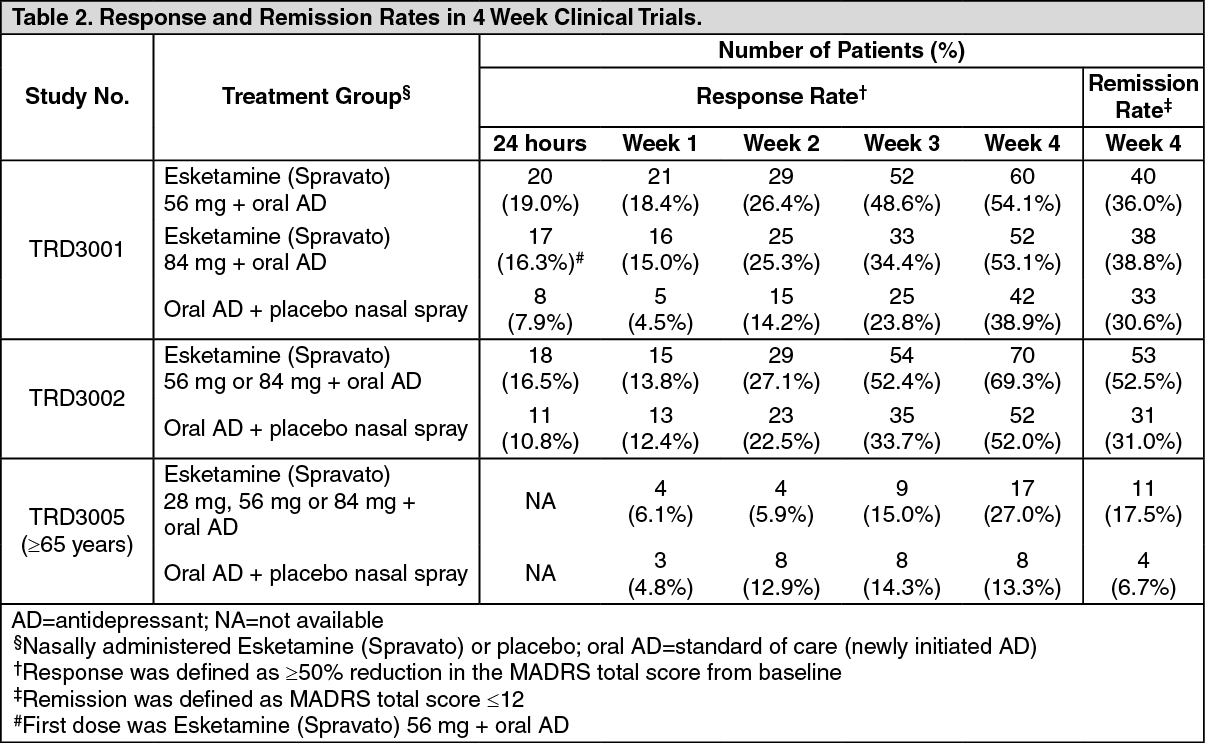
Click on icon to see table/diagram/imageResponse and remission rates: Response was defined as ≥50% reduction in the MADRS total score from baseline of the induction phase. Based on the reduction in MADRS total score from baseline, the proportion of patients in Studies TRD3001, TRD3002 and TRD3005 who demonstrated response to Esketamine (Spravato) plus oral AD treatment was greater than for oral AD plus placebo nasal spray throughout the 4‑week double‑blind induction phase (Table 2).
Remission was defined as a MADRS total score ≤12. In all three studies, a greater proportion of patients treated with Esketamine (Spravato) plus oral AD were in remission at the end of the 4‑week double‑blind induction phase than for oral AD plus placebo nasal spray (Table 2). (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageTreatment‑resistant depression - Long‑term studies: Relapse‑prevention study: Study SUSTAIN‑1 (TRD3003) was a long‑term randomized, double‑blind, parallel‑group, active‑controlled, multicenter, relapse prevention study. Overall a total of 705 patients were enrolled; 437 directly enrolled; 150 transferred from TRD3001, and 118 transferred from TRD3002. Patients directly enrolled were administered Esketamine (Spravato) (56 mg or 84 mg twice weekly) plus oral AD in a 4‑week open label induction phase. Patients who were responders (MADRS total score reduction ≥50% from baseline), continued receiving treatment with Esketamine (Spravato) plus oral AD in a 12‑week optimization phase. At the end of the open label induction phase, 52% of patients were in remission (MADRS total score ≤12) and 66% of patients were responders (≥50% improvement in MADRS total score). Four hundred fifty‑five (455) esketamine‑treated patients entered the optimization phase, patients in stable remission or stable response were randomized to continue with Esketamine (Spravato) or stop Esketamine (Spravato) and switch to placebo nasal spray. After an initial 16 weeks of treatment with Esketamine (Spravato) plus oral AD, 176 (39%) patients were in stable remission and 121 (27%) patients were in stable response (but not in stable remission). Stable remission was defined as MADRS total score ≤12 in at least 3 of the last 4 weeks of the optimization phase and stable response was defined as ≥50% reduction in the MADRS total score from baseline for the last 2 weeks of the optimization phase, but not in stable remission.
The baseline demographic and disease characteristics of the patients randomized to the double‑blind maintenance phase were similar between the Esketamine (Spravato) plus oral AD and oral AD plus placebo groups, median patient age was 48 years (range 19 to 64 years), 66% were female; 90% Caucasian and 4% of African descent.
Stable Remission: Patients in stable remission who continued treatment with Esketamine (Spravato) plus oral AD experienced a statistically significantly longer time to relapse of depressive symptoms than did patients on standard of care (oral AD) plus placebo nasal spray (Figure 3). Relapse was defined as a MADRS total score ≥22 for 2 consecutive weeks or hospitalization for worsening depression or any other clinically relevant event indicative of relapse. The median time to relapse for standard of care (oral AD) plus placebo nasal spray group was 273 days, whereas the median was not estimable for Esketamine (Spravato) plus oral AD, as this group never reached 50% relapse rate. (See Figure 3.)
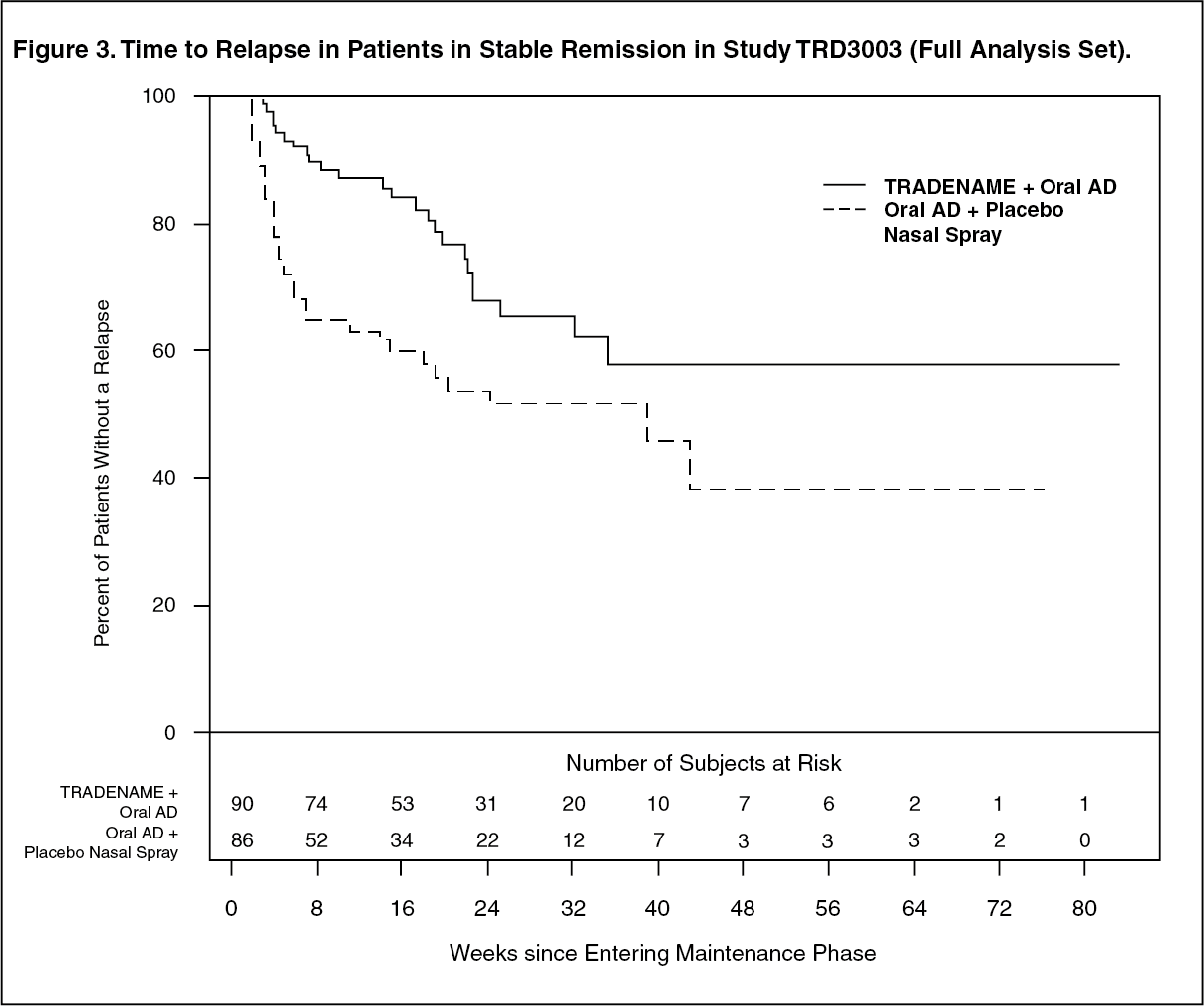 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageFor patients in stable remission, the estimated hazard ratio (95% CI) of Esketamine (Spravato) plus oral AD relative to standard of care (oral AD) plus placebo nasal spray based on weighted estimates was 0.49 (95% CI: 0.29, 0.84), indicating that, patients who were in stable remission and continued treatment with Esketamine (Spravato) plus oral AD group were on average 51% less likely to relapse than patients who switched to standard of care (oral AD) plus placebo nasal spray.
Stable Response: The efficacy results were also consistent for patients in stable response who continued treatment with Esketamine (Spravato) plus oral AD; patients experienced a statistically significantly longer time to relapse of depressive symptoms than did patients on standard of care (oral AD) plus placebo nasal spray (Figure 4). The median time to relapse for standard of care (oral AD) plus placebo nasal spray group (88 days) was shorter compared to Esketamine (Spravato) plus oral AD group (635 days). (See Figure 4.)
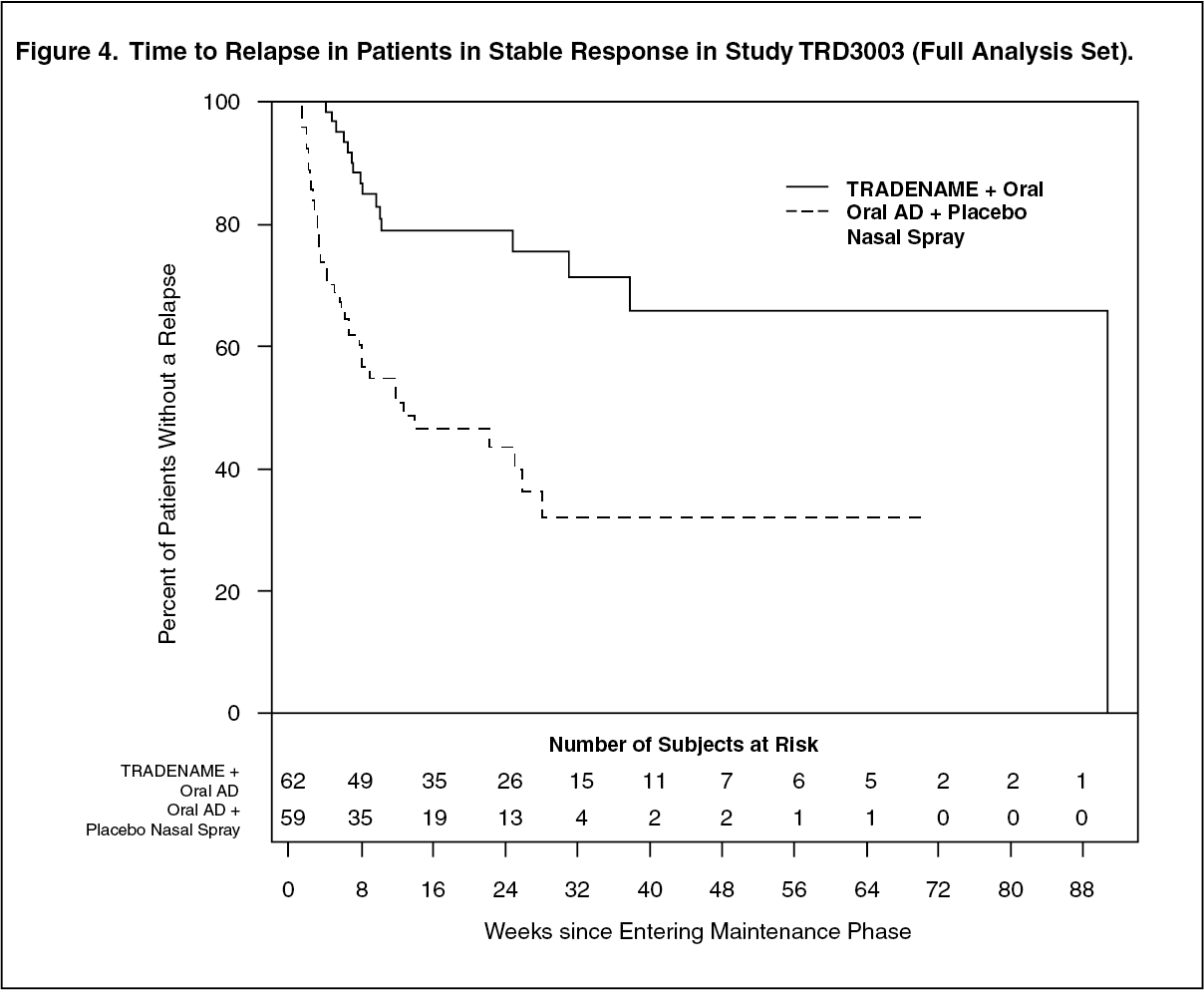 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageFor patients in stable response, the estimated hazard ratio (95% CI) of Esketamine (Spravato) plus oral AD relative to standard of care (oral AD) plus placebo nasal spray based on Cox proportional hazards model was 0.30 (95% CI: 0.16, 0.55), indicating that, patients who were stable responders and continued treatment with Esketamine (Spravato) plus oral AD group were on average 70% less likely to have a relapse than patients who switched to standard of care (oral AD) plus placebo nasal spray.
Enrollment in TRD3003 was staggered over approximately 2 years. The maintenance phase was of variable duration and continued until the individual patient had a relapse of depressive symptoms or discontinued for any other reason, or the study ended because the required number of relapse events occurred. Exposure numbers were influenced by the study stopping at a pre‑determined number of relapses based on the interim analysis. After an initial 16 weeks of treatment with Esketamine (Spravato) plus oral AD, the median duration of exposure to Esketamine (Spravato) in the maintenance phase was 4.2 months (range: 1 day to 21.2 months) in Esketamine (Spravato)‑treated patients (stable remission and stable response). In this study, 31.6% of patients received Esketamine (Spravato) for greater than 6 months and 7.9% of patients received Esketamine (Spravato) for greater than 1 year in the maintenance phase.
Dosing Frequency: Starting from week 8, an algorithm (based on the MADRS) was used to determine the dosing frequency; patients in remission (i.e., MADRS total score was ≤12) were dosed every other week, however, if the MADRS total score increased to >12, then the frequency was increased to weekly dosing for the next 4 weeks; with the objective of maintaining the patient on the lowest dosing frequency to maintain response/remission. The dosing frequency used the majority of the time during the maintenance phase is shown in Table 3. Of the patients randomized to Esketamine (Spravato), 60% received 84 mg and 40% received 56 mg dose. (See Table 3.)
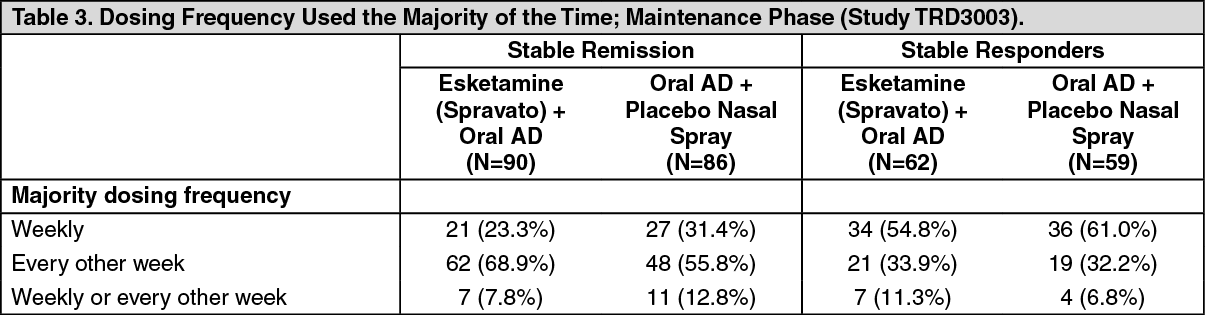 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageOpen‑label Long‑term Safety and Efficacy Study: Study SUSTAIN‑2 (TRD3004) was an open‑label, long‑term study of Esketamine (Spravato) plus oral AD in patients with TRD.
The primary objective was to evaluate the long‑term (up to 52 weeks) safety and efficacy of Esketamine (Spravato). Esketamine (Spravato) was not associated with effects on cognitive function or treatment‑emergent symptoms of interstitial cystitis. In the elderly subgroup (≥65 years of age) slowing of reaction time starting at Week 20 and through the end of the study was observed, however, performance on other cognitive tests remained stable.
In addition, there was no evidence of withdrawal and/or rebound symptoms following cessation of Esketamine (Spravato) treatment. No cases of respiratory depression were reported and there was no evidence of treatment related changes in lab parameters.
Mean body weight remained stable during treatment with Esketamine (Spravato) plus oral AD both in the induction phase and maintenance phase (mean change from baseline ± standard deviation of ‑0.29±2.15 kg at Day 28 and 0.44±5.83 kg at Week 48).
TRD3004 also evaluated long‑term efficacy, including effects on depressive symptoms. At the end of the 4‑week induction phase, the response rate (≥50% improvement from Baseline in the MADRS total score) was 78.4% (593/756) and remission rate (MADRS total score ≤12) was 47.2% (357/756); of the responders proceeding to the maintenance phase, 76.5% (461/603) were in response and 58.2% (351/603) were in remission at endpoint.
Dose‑response study in treatment‑resistant depression: A Phase 2 adjunctive, doubly‑randomized, double‑blind, placebo‑controlled, dose‑ranging study, enrolled 108 adult patients with TRD. Adjunctive to continued oral AD therapy, patients were treated with esketamine 14 mg, 28 mg, 56 mg or 84 mg or placebo administered nasally twice a week for 2 weeks. Treatment with the 28‑mg, 56‑mg and 84‑mg doses of Esketamine (Spravato) significantly improved depressive symptoms in patients with TRD as demonstrated by the change in MADRS total score after 1 week. While Esketamine (Spravato) doses of 28 mg, 56 mg and 84 mg were efficacious in TRD treatment, the duration of the efficacy of the 28‑mg dose was shorter.
Response rates at Day 8 of Period 1 for the double‑blind phase are shown as follows (Table 4). (See Table 4.)
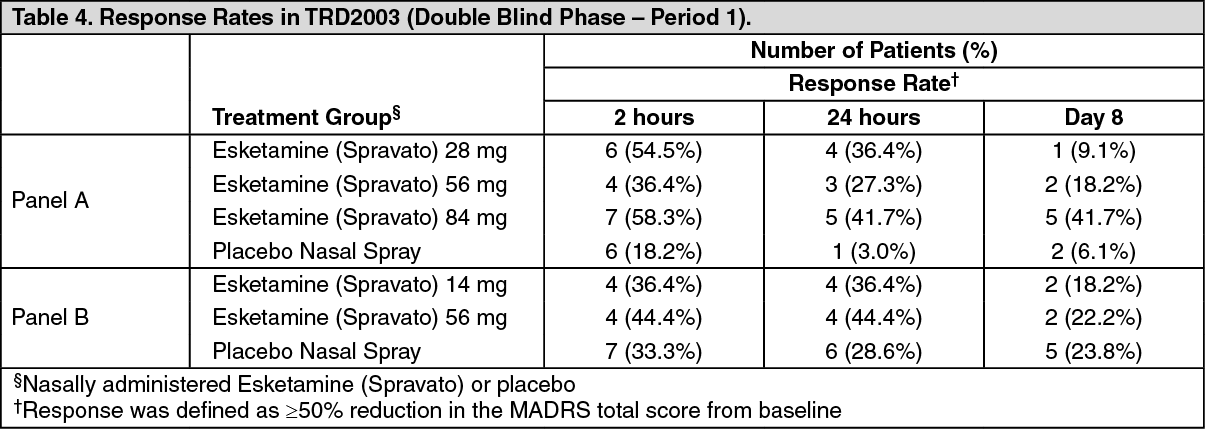 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageMajor Depressive Disorder with acute suicidal ideation or behavior: Esketamine (Spravato) was evaluated in two identical Phase 3 short term (4 week) randomized, double blind, multicenter, placebo controlled studies, Aspire I (SUI3001; NCT03039192) and Aspire II (SUI3002; NCT03097133) in adult patients with moderate to severe MDD (MADRS total score >28) who had active suicidal ideation with intent. In these studies, patients received treatment with Esketamine (Spravato) 84 mg or placebo nasal spray twice weekly for 4 weeks. All patients received comprehensive standard of care (SOC) treatment, including an initial inpatient hospitalization and a newly initiated or optimized oral antidepressant (AD) therapy (AD monotherapy or AD plus augmentation) as determined by the investigator. After the first dose, a one-time dose reduction to Esketamine (Spravato) 56 mg was allowed for patients unable to tolerate the 84 mg dose.
The baseline demographic and disease characteristics of patients in SUI3001 and SUI3002 were similar between the Esketamine (Spravato) plus SOC or placebo nasal spray plus SOC groups. The median patient age was 40 years (range 18 to 64 years), 61% were female; 73% Caucasian and 6% Black; and 63% of patients had at least one prior suicide attempt. Prior to entering the study, 92% of the patients were receiving antidepressant therapy. During the study, as part of standard of care treatment, 40% of patients received AD monotherapy, 54% of patients received AD plus augmentation regimen, and 6% received both AD monotherapy/AD plus augmentation regimen.
The primary efficacy measure was the reduction of symptoms of MDD as measured by the change from baseline MADRS total score at 24 hours after first dose (Day 2).
In SUI3001 and SUI3002, Esketamine (Spravato) plus SOC demonstrated statistical superiority on the primary efficacy measure compared to placebo nasal spray plus SOC (see Table 5).
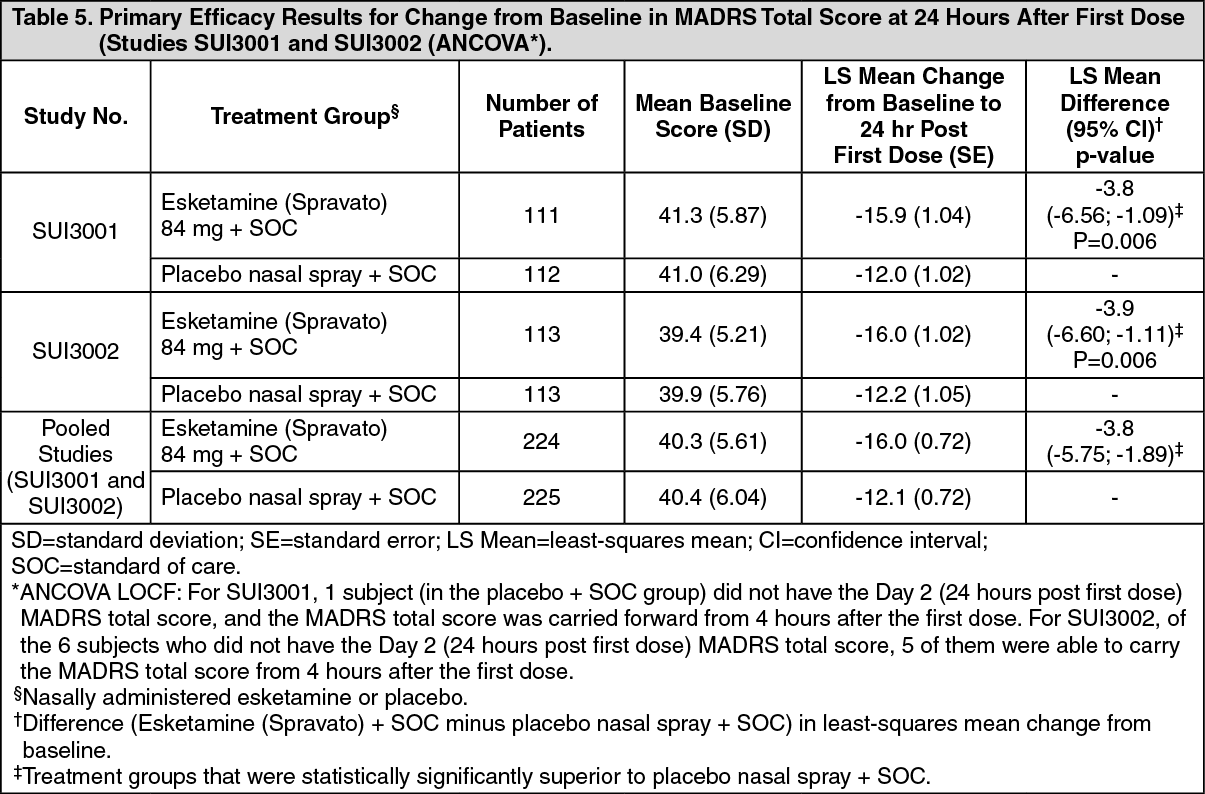 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe treatment differences (95% CI) in change from baseline in MADRS total score at Day 2 (24 hours post first dose) between Esketamine (Spravato) + SOC and placebo + SOC were 4.81 (7.26; 2.36) for the subpopulation that reported a prior suicide attempt (N=282) and 2.32 (5.54; 0.91) for the subpopulation that did not report a prior suicide attempt (N=166).
Time Course of Treatment Response: In both SUI3001 and SUI3002, Esketamine (Spravato)'s treatment difference compared to placebo was observed starting at 4 hours. Between 4 hours and Day 25, both the Esketamine (Spravato) and placebo groups continued to improve; the difference between the groups generally remained but did not appear to increase over time through Day 25. Figure 5 depicts time course of the primary efficacy measure of change in MADRS total score using pooled SUI3001 and SUI3002. (See Figure 5.)
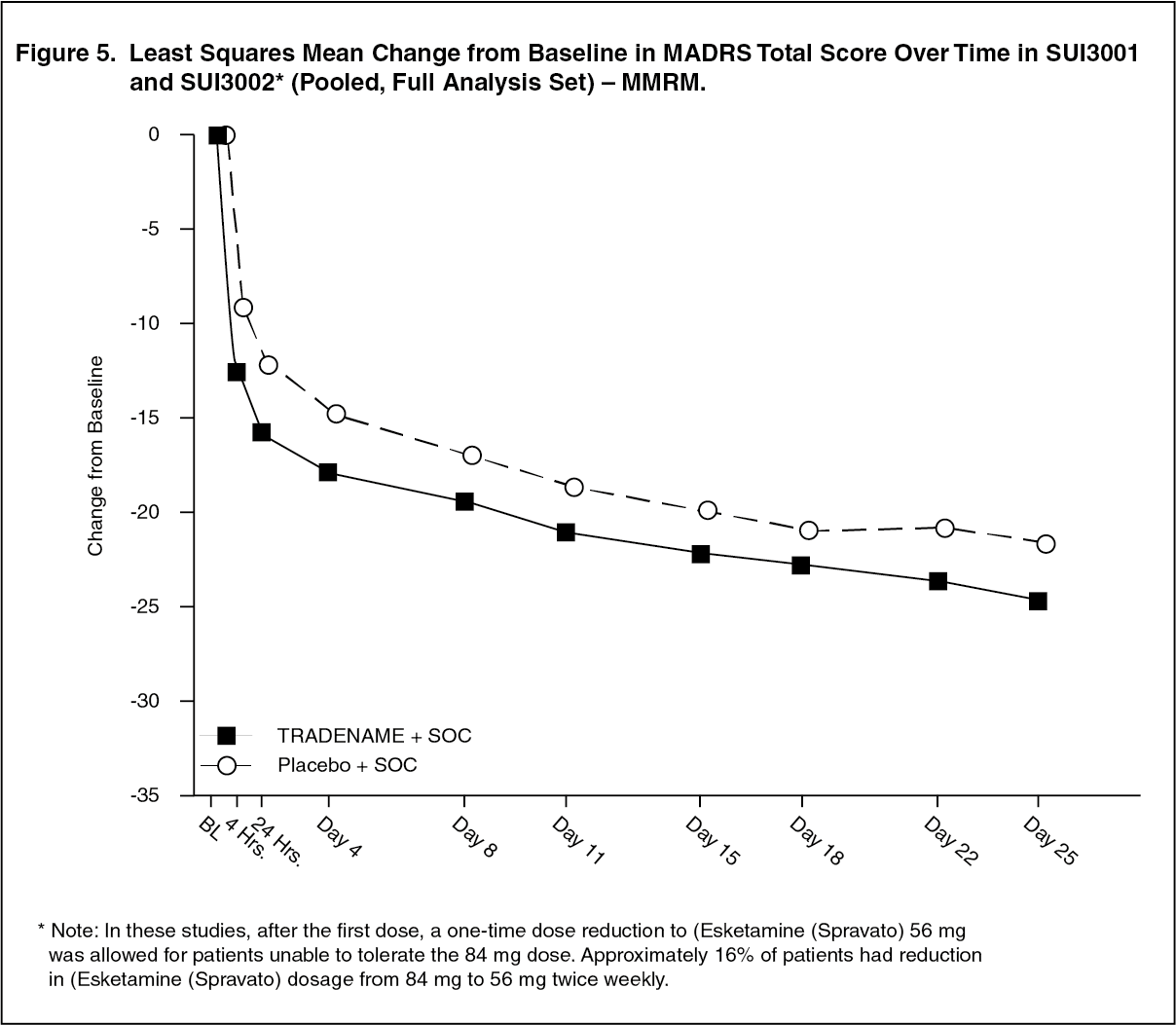 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageRemission rates: In the Phase 3 studies, the percentage of patients who achieved remission (MADRS total score ≤12 at any given time during the study) was greater in the Esketamine (Spravato) + SOC group than in the placebo + SOC group at all timepoints during the double-blind treatment phase (Table 6). (See Table 6.)
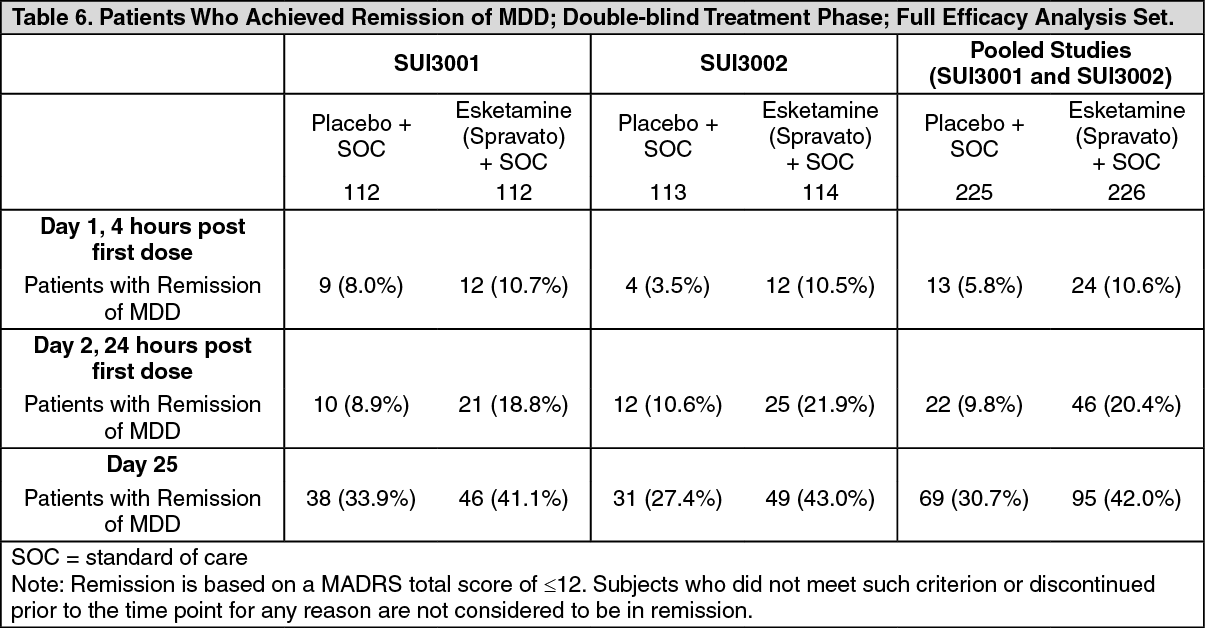 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageEffects on Suicidality: Overall patients in both treatment groups experienced improvement in the severity of their suicidality as measured by the Clinical Global Impression - Severity of Suicidality - Revised (CGI-SS-r) scale at the 24-hour endpoint, though there was no statistically significant difference between treatment groups. The long-term efficacy of Esketamine (Spravato) to prevent suicide has not been established.
Pharmacokinetics: Absorption: The mean absolute bioavailability of 84 mg esketamine administered as a nasal spray is approximately 48%.
Esketamine is rapidly absorbed by the nasal mucosa following nasal administration and can be measured in plasma within 7 minutes following a 28‑mg dose. The time to reach maximum plasma concentration (tmax) is typically 20 to 40 minutes after the last nasal spray of a treatment session (see Dosage & Administration).
Dose‑dependent, linear increases in the plasma Cmax and AUC∞ of esketamine nasal spray were produced by doses of 28 mg, 56 mg and 84 mg.
The pharmacokinetic profile of esketamine is similar after a single dose and repeat dose administration with no accumulation in plasma when esketamine is administered twice a week.
Distribution: The mean steady‑state volume of distribution of esketamine administered by the intravenous route is 709 L.
The proportion of the total concentration of esketamine that is bound to proteins in human plasma is on average 43 to 45%. The degree to which esketamine is bound to plasma proteins is not dependent on hepatic or renal function.
Esketamine is not a substrate of transporters P‑glycoprotein (P‑gp; multidrug resistance protein 1), breast cancer resistance protein (BCRP), or organic anion transporter (OATP) 1B1, or OATP1B3. Esketamine does not inhibit these transporters or multi‑drug and toxin extrusion 1 (MATE1) and MATE2‑K, or organic cation transporter 2 (OCT2), OAT1, or OAT3.
Metabolism: Esketamine is extensively metabolized in the liver. The primary metabolic pathway of esketamine in human liver microsomes is N‑demethylation to form noresketamine. The main CYP enzymes responsible for esketamine N‑demethylation are CYP2B6 and CYP3A4. Other CYP enzymes, including CYP2C19 and CYP2C9, contribute to a much smaller extent. Noresketamine is subsequently metabolized via CYP‑dependent pathways to other metabolites, some of which undergo glucuronidation.
Excretion: The mean clearance of esketamine administered by the intravenous route was approximately 89 L/hour. After Cmax was reached following nasal administration, the decline in esketamine concentrations in plasma was rapid for the first few hours and then more gradual. The mean terminal half‑life following administration as a nasal spray generally ranged from 7 to 12 hours.
Following intravenous administration of radiolabelled esketamine, approximately 78% and 2% of administered radioactivity was recovered in urine and feces, respectively. Following oral administration of radiolabelled esketamine, approximately 86% and 2% of administered radioactivity was recovered in urine and feces, respectively. The recovered radioactivity consisted primarily of esketamine metabolites. For the intravenous and oral routes of administration, <1% of the dose was excreted in the urine as unchanged drug.
Special populations: Elderly (65 years of age and older): The pharmacokinetics of esketamine administered as a nasal spray was compared between elderly but otherwise healthy subjects and younger healthy adults. The mean esketamine Cmax and AUC∞ values produced by a 28‑mg dose were 21% and 18% higher, respectively, in elderly subjects (age range 65 to 81 years) compared with younger adult subjects (age range 22 to 50 years). The mean esketamine Cmax and AUC∞ values produced by an 84‑mg dose were 67% and 38% higher, respectively, in elderly subjects (age range 75 to 85 years) compared with younger adult subjects (age range 24 to 54 years). The terminal half‑life of esketamine was similar in the elderly and younger adult subjects.
Renal impairment: Relative to the subjects with normal renal function (creatinine clearance [CLCR], 88 to 140 mL/min), the Cmax of esketamine was on average 20 to 26% higher in subjects with mild (CLCR, 58 to 77 mL/min), moderate (CLCR, 30 to 47 mL/min), or severe (CLCR, 5 to 28 mL/min, not on dialysis) renal impairment following administration of a 28‑mg dose of esketamine nasal spray. The AUC∞ was 13 to 36% higher in the subjects with mild to severe renal impairment.
There is no clinical experience with esketamine administered as a nasal spray in patients on dialysis.
Hepatic impairment: The Cmax and AUC∞ of esketamine produced by a 28‑mg doses were similar between subjects with Child‑Pugh class A (mild) hepatic impairment and healthy subjects. The Cmax and AUC∞ of esketamine were 8% higher and 103% higher, respectively, in subjects with Child‑Pugh class B (moderate) hepatic impairment, relative to healthy subjects.
There is no clinical experience with esketamine administered as a nasal spray in patients with Child‑Pugh class C (severe) hepatic impairment.
Race: The pharmacokinetics of esketamine nasal spray was compared between healthy Asian subjects and Caucasian subjects. Mean plasma esketamine Cmax and AUC∞ values produced by a single, 56‑mg dose of esketamine were approximately 14% and 33% higher, respectively, in Chinese subjects compared to Caucasians. Both parameters were approximately 40% higher in Japanese subjects, relative to Caucasian subjects. On average, esketamine Cmax was 10% lower and AUC∞ was 17% greater in Korean subjects, relative to Caucasian subjects. The mean terminal half‑life of esketamine in the plasma of Asian subjects ranged from 7.1 to 8.9 hours and was 6.8 hours in Caucasian subjects.
Gender: A population pharmacokinetic analysis was conducted that included healthy subjects (138 males and 118 females) and patients with major depressive disorder (203 males and 361 females). The results indicated that the pharmacokinetics of esketamine administered as a nasal spray is not influenced by gender.
Body Weight: A population pharmacokinetic analysis was conducted that included 256 healthy subjects and 564 patients with major depressive disorder. The total body weight of the subjects ranged from 39 to 170 kg. The results indicated that the pharmacokinetics of esketamine administered as a nasal spray is not influenced by body weight.
Allergic rhinitis: The pharmacokinetics of a single, 56‑mg dose of esketamine administered as a nasal spray was similar in subjects with allergic rhinitis who were exposed to grass pollen compared to healthy subjects.
Toxicology: Non-clinical Information: General Toxicity: Once‑daily nasal administration of esketamine in rats up to 9 mg/day for 6 months, and dogs up to 72 mg/day for 9 months, resulted in non‑adverse central nervous system‑related clinical signs reflecting the anesthetic properties of the test article. No notable lesions were found in the nasal cavity or any peripheral organ. After 3 months of daily treatment at 9 mg/day in rats, the systemic exposure of esketamine (Cmax and AUC) resembled that in humans at the maximum recommended human dose (MRHD) of 84 mg, while the Cmax‑ and AUC‑based exposure ratios for esketamine in dogs after 3 months of daily treatment at 72 mg/day were approximately 4‑ and 1‑fold, respectively.
Neurotoxicity: In single‑dose and 14‑day repeated‑dose neurotoxicity studies with nasally‑administered esketamine in rats, no histopathological brain lesions were noted. In single dose neurotoxicity studies, where rats were nasally‑administered with esketamine at a dose up to 72 mg, the Cmax‑ and AUC‑based safety margins for esketamine were approximately 59‑ and 86‑fold, respectively, compared to the human exposure at the MRHD of 84 mg. In a 14‑day neurotoxicity study where rats received nasally‑administered esketamine once daily up to a dose of 54 mg/day, the Cmax‑ and AUC‑based safety margins for esketamine were approximately 17‑ and 11‑fold. Moreover, no evidence of neurotoxicity was found in the 6‑month rat and the 9‑month dog repeated‑dose toxicology studies with once daily nasal administration of esketamine as judged by brain histopathology and functional assessments. Similarly, no neurotoxicity was noted in the shorter‑term animal toxicology studies with nasally‑administered esketamine. Overall, the risk of neurotoxicity associated with nasal administration of esketamine to patients is expected to be low.
Carcinogenicity and Mutagenicity: Once‑daily nasal administration of esketamine did not increase the incidence of tumors in a 2‑year rat carcinogenicity study at doses up to 9 mg/day. At this dose, the exposure to esketamine resembled the human exposure at the MRHD of 84 mg. Esketamine was not carcinogenic either upon once‑daily subcutaneous administration in a 6‑month study in transgenic (Tg.rasH2) mice at doses up to 70/40 mg/kg/day. At that dose, the Cmax‑ and AUC‑based exposure ratios for esketamine were approximately 20‑ and 4‑fold, respectively, compared to the MRHD of 84 mg.
Esketamine was not mutagenic with or without metabolic activation in the Ames test. Genotoxic effects with esketamine were seen in a screening in vitro micronucleus test in the presence of metabolic activation. However, intravenously‑administered esketamine was devoid of genotoxic properties in an in vivo bone marrow micronucleus test in rats and an in vivo Comet assay in rat liver cells. In simulated gastric fluid there is no evidence that N‑nitroso‑esketamine is formed out of the fraction of the nasally‑administered dose of esketamine that is orally absorbed.
Reproductive Toxicity: In an embryo‑fetal developmental toxicity study with nasally‑administered ketamine in rats, the offspring was not adversely affected in the presence of maternal toxicity at doses up to 150 mg/kg/day. In rats, the Cmax- and AUC‑based safety margin estimated for esketamine at the 150 mg/kg/day dose of ketamine was 61‑ and 12‑fold compared to the maximum recommended human dose (MRHD) of esketamine of 84 mg. In pregnant rabbits, racemic ketamine was administered intranasally from gestational day 6 to 18 at doses of 10, 30, and 100 mg/kg/day. The high dose was lowered from 100 to 50 mg/kg after 5 days of dosing due to excessive mortality in the pregnant rabbits. Skeletal malformations were observed at doses ≥ 30 mg/kg/day, which were maternally toxic. In rabbits, the estimated exposure to esketamine at the 10 mg/kg/day no‑effect dose of ketamine was below the maximum exposure to esketamine at 84 mg in humans.
Animal studies with ketamine showed evidence of developmental neurotoxicity. The potential for esketamine to have neurotoxic effects on developing fetuses cannot be excluded. (See Use in Pregnancy & Lactation).
In a pre‑ and postnatal developmental toxicity study with nasally‑administered esketamine up to 9 mg/day in rats, no adverse effects occurred in the dams nor their offspring.
Fertility: In a fertility and early embryonic developmental toxicity study, esketamine nasally‑administered to rats at 0.9, 3, or 9 mg/day caused maternal and paternal toxicity at 3 and 9 mg/day. Fertility and reproductive capacities were not adversely affected at any dose.