Pharmacology: Mechanism of Action: Xyntha, recombinant coagulation factor VIII, is a glycoprotein with an approximate molecular mass of 170,000 Da, consisting of 1,438 amino acids, which does not contain the non-functional B-domain.
Xyntha is a recombinant DNA-based substance which has functional characteristics comparable to those of endogenous factor VIII. Activated factor VIII acts as a cofactor for activated factor IX accelerating the conversion of factor X to activated factor X. Activated factor X converts prothrombin into thrombin.
Thrombin then converts fibrinogen into fibrin which forms an insoluble clot.
The factor VIII/von Willebrand factor complex consists of two molecules (factor VIII and von Willebrand factor) with different physiological functions. When infused into a patient with hemophilia, factor VIII binds to von Willebrand factor in the patient's circulation.
Hemophilia A is an X chromosome-linked hereditary disorder of blood coagulation due to decreased levels of factor VIII:C and results in bleeding into joints, muscles or internal organs, either spontaneously or as a result of accidental or surgical trauma. Factor VIII activity is greatly reduced in patients with hemophilia A. The administration of Xyntha increases plasma levels of factor VIII activity and can temporarily correct the coagulation defect in these patients.
Pharmacodynamics: Factor VIII is the specific clotting factor deficient in patients with hemophilia A (classical hemophilia). The administration of Xyntha, Antihemophilic Factor (Recombinant)[BDDrFVIII] increases plasma levels of factor VIII and can temporarily correct the coagulation defect in these patients.
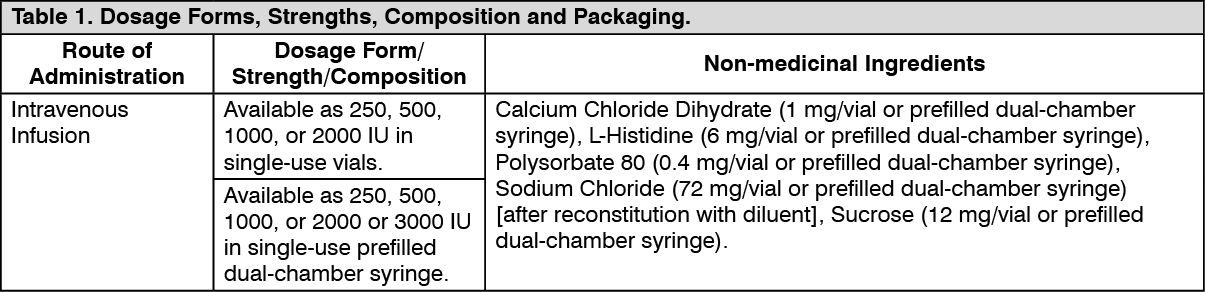
Clinical Trials: Clinical Trials by Indication: The following table lists the registrational studies conducted with ReFacto and ReFacto AF or XYNTHA. (See Table 2.)
 Click on icon to see table/diagram/image
Study Results:
Click on icon to see table/diagram/image
Study Results: In a pivotal phase 3 study (study 310), the efficacy of Xyntha was evaluated in routine prophylaxis and on-demand treatment. Prophylaxis was to be initiated at a dose of 30 IU/kg given 3 times per week. The on-demand treatment dosing regimen was to be determined by the investigator. Ninety-four (94) PTPs with moderately severe or severe hemophilia A (FVIII:C ≤2%) received at least 1 dose of Xyntha and were included in the intent-to-treat (ITT) population. Eighty-nine (89) patients accrued at least 50 exposure days (EDs) to Xyntha in the study.
Of the 94 patients in the ITT population, 30 patients with FVIII:C ≤1% also participated in the double-blind, randomized, crossover PK period of the study and were included in the per-protocol population for analyses of bioequivalence versus another rFVIII product, Advate, and full pharmacokinetic characterization. Both endpoints were surrogate markers for clinical efficacy. The results of these analyses showed that Xyntha is bioequivalent to Advate and the pharmacokinetic profile of Xyntha remained stable after 6 months of repeated use.
Intent-to-treat analysis of clinical efficacy variables in the open-label safety and efficacy period yielded similarly positive outcomes. All 94 patients received Xyntha for routine prophylaxis; the median dose administered was 30.2 IU/kg (range, 6.8 to 76.9 IU/kg). Most patients (57/94; 60.6%) reported no spontaneous bleeding while on routine prophylaxis. The median annualized bleeding rate (ABR) for all bleeding episodes was 1.9 (mean 3.9, range 0 to 42.1), indicating effective prevention of bleeding in the study population. Fifty-three (53) of 94 patients received Xyntha for on-demand treatment; the median dose administered was 30.6 IU/kg (range, 6.4 to 74.4 IU/kg). The majority of bleeding episodes (173/187; 92.5%) resolved with 1 or 2 infusions. This outcome was not restricted to any particular bleeding location as similar efficacy was seen in bleeding occurring in joints, soft tissues/muscles, and other sites. A wide range of doses was used to initiate treatment of bleeding; however, the distribution of doses used to initiate treatment of bleeding was similar regardless of location of bleeding. Patients rated the majority of infusions used to initiate treatment of bleeding as either excellent or good (132/187; 70.6%). The incidence of less than expected therapeutic effect (LETE) occurred at a rate of 0.4% (25/6404 prophylactic infusions) when Xyntha was administered for prophylaxis and 0.5% (1/187 bleeding episodes) when administered for on-demand treatment.
In another series of clinical trials (studies 300 and 301), the efficacy of Xyntha manufactured by the previous process was evaluated in uncontrolled phase 3 studies of 113 PTPs and 101 PUPs who received Xyntha manufactured by the previous process for on-demand treatment, routine prophylaxis, and/or surgical prophylaxis and were followed for up to 6 years. Hemostatic efficacy was rated on an ordinal scale of excellent, good, fair, and none.
In 112 of 113 PTPs treated on-demand, a total of 10,882 bleeding episodes were reported, with a median of 77.5 bleeding episodes per study subject. Of these, the hemostatic efficacy of Xyntha manufactured by the previous process was assessed following the first infusion for treatment of 10,445 bleeding episodes: 9944 (95%) were rated excellent or good in their response to treatment, 429 (4%) were rated fair, and 72 (0.7%) were rated as having no response; 4% (437/10,882) of the bleeding episodes were not rated. Of the 10,882 bleeding episodes, 7981 (73%) were managed with a single infusion, 1612 (15%) required 2 infusions, 623 (6%) required 3 infusions, and 666 (6%) required 4 or more infusions for satisfactory resolution. The mean dose per infusion was 31 IU/kg.
In 100 of 101 PUPs treated on-demand, a total of 2715 bleeding episodes were reported with a median of 19.5 bleeding episodes per study subject. Of these, the hemostatic efficacy of Xyntha manufactured by the previous process was assessed following the first infusion for treatment of 2604 bleeding episodes: 2459 (94%) were rated excellent or good in their response to treatment, 142 (5%) were rated fair, and 3 (0.1%) were rated as having no response; 4% (111/2,715) of the bleeding episodes were not rated. Of the 2715 bleeding episodes, 1794 (66%) were managed with a single infusion, 502 (19%) required 2 infusions, 229 (8%) required 3 infusions, and 190 (7%) required 4 or more infusions for satisfactory resolution. The mean dose per infusion was 51 IU/kg.
Xyntha manufactured by the previous process has been studied in short-term routine prophylaxis. In uncontrolled phase 3 clinical trials, a mean dose of 27 ± 11 IU/kg per infusion in PTPs (n=85) and a mean dose of 49 ± 17 IU/kg per infusion in PUPs (n=45) was given repeatedly at variable intervals (for PTPs: median 94 weeks, range 3-296 weeks; for PUPs: median 61 weeks, range 2-222 weeks). In PTPs and PUPs, the mean rate of spontaneous musculoskeletal bleeding episodes was lower during periods of routine prophylaxis. PTPs (n=85) had a mean of 10 bleeding episodes (spontaneous and injury-related) per year during the prophylactic periods compared to a mean of 25 bleeding episodes per year during on-demand periods. PUPs (n=45) had a mean of 6 bleeding episodes (spontaneous and injury-related) per year during the prophylactic periods compared to a mean of 11 bleeding episodes per year during the on-demand periods. These non-randomized trial results should be interpreted with caution, as the investigators exercised their own discretion in deciding when and in whom prophylaxis was to be initiated and terminated.
A pivotal phase 3 study (Study 311) for surgical prophylaxis in patients with hemophilia A included PTPs with severe or moderately severe (FVIII:C ≤2%) hemophilia A undergoing major surgical procedures who received Xyntha. Thirty (30) patients were treated with Xyntha and comprised the ITT population; 29 patients underwent major surgery and completed the study. Thirty (30) subjects were assigned to receive Xyntha by bolus injection (BI; 22 patients) or by continuous infusion (CI; 8 patients) at the health professional's discretion to support surgical hemostasis followed by inpatient and outpatient postoperative care. One subject assigned to CI received Xyntha for a pre-surgery pharmacokinetic assessment only and subsequently elected not to undergo surgery. The 22 patients treated by BI received a total of 942 infusions (ranging from 16 to 72 infusions per patient) for a cumulative total dose of 2,037,386 IU of Xyntha over 682 cumulative total exposure days (EDs) (ranging from 15 to 40 EDs per patient). The 8 patients assigned to treatment by CI, including 1 patient who received only 1 dose for PK assessment, received a total dose of 529,977 IU of Xyntha over 204 total EDs (range 1 to 37 EDs per patient).
Of the 29 patients who underwent surgery, 25 were included in the efficacy evaluable population. Major surgical procedures for the 25 efficacy evaluable subjects were 11 total knee replacements, 1 hip replacement, 5 synovectomies, 1 left ulnar nerve transposition release, 1 ventral hernia repair/scar revision, 1 knee arthroscopy, 1 revision and debridement of the knee after a total knee replacement, 1 hip arthroplasty revision, 1 stapes replacement, 1 ankle arthrodesis, and 1 pseudotumor excision. For the 25 surgical subjects, investigator's ratings of the efficacy at the end of surgery were excellent for 72% (18/25) and good for 28% (7/25) of patients and at the end of the initial postoperative period were excellent for 92% (23/25) and good for 8% (2/25) of patients. Intraoperative blood loss was reported as normal or absent for all procedures. Thirteen of the 25 evaluable patients had blood loss in the postoperative period, and in 10 cases the postoperative blood loss was rated normal. In 3 cases, the postoperative blood loss was rated abnormal: 1 due to hemorrhage following surgical trauma to the epigastric artery, 1 due to an 800 mL blood loss after hip replacement surgery, and 1 after an elbow synovectomy where the blood loss could not be measured by the investigator.
In a supporting study of Xyntha, there were 6 surgical procedures that were classified as major per the definitions of the pivotal surgery study. In all cases, hemostatic efficacy was effectively managed with Xyntha. No patient had blood loss greater than 50 mL, and no blood transfusions were given.
Data in pediatric population <16 years of age: In study 313, the safety and efficacy of XYNTHA, and FVIII:C pharmacokinetics after XYNTHA infusion in children <16 years of age with moderately severe to severe hemophilia A (FVIII:C ≤2%) were evaluated in an open-label study. The study (1) compared the efficacy of routine prophylaxis to on-demand treatment in a cohort of pediatric subjects <6 years of age, and (2) compared two routine prophylaxis regimens in a cohort of children <16 years of age (25 IU/kg every other day (EOD) versus 45 IU/kg bi-weekly (BIW)).
Fifty-one (51) subjects with at least 20 prior EDs to FVIII products were enrolled and included in the ITT population. Fifty (50) subjects received at least 1 dose of Xyntha, and 41 subjects completed the study. Nine (9) pediatric subjects <6 years of age (1 of whom did not receive any treatment dose) received on-demand treatment with Xyntha at a median dose of 24 IU/kg for a 6-month period followed by a routine prophylaxis regimen at a dose of 25 IU/kg every other day (EOD) for 12 months. The median ABR observed during the on-demand treatment period was 34.0 (mean 47.0, range 0 to 92.4) compared to 0.6 (mean 1.5, range 0 to 6.2) while on the routine prophylaxis regimen (p=0.0040).
Forty-two (42) pediatric subjects <16 years of age received a routine prophylaxis dosing regimen of either 45 IU/kg BIW or 25 IU/kg EOD for 12 months before crossing over to receive the alternate regimen, and 35 subjects provided data for both regimens. Because the 90% confidence interval (CI) for the difference of [(0.03, 2.22)] was inside the prospectively defined equivalence limit of (-3.3), equivalent efficacy was established with respect to ABR for both regimens (mean ± SD 3.3±5.3 compared to 2.2±4.1).
Pharmacokinetics: One Stage Assay: In a pivotal cross-over pharmacokinetic study, Xyntha was shown to be bioequivalent to another recombinant factor VIII product (rFVIII, Advate) in 30 previously treated patients (PTPs) (≥ 12 years) using the one-stage clotting assay. The ratios of geometric least square means of Xyntha-to-Advate were 100%, 89.8% and 88.0% for K-value, AUC
t and AUC
∞, respectively. The corresponding 90% confidence intervals about the ratios of Xyntha to Advate geometric means were within the bioequivalence window of 80% to 125%, demonstrating bioequivalence of Xyntha to Advate. (See Figure.)
Click on icon to see table/diagram/image
In the same study, the pharmacokinetic parameters for Xyntha were determined at baseline and followed-up in 25 PTPs (≥12 years) after repeated administration of Xyntha for six months. At baseline, following a single 2-minute intravenous infusion of 50 IU/kg dose of Xyntha, plasma FVIII:C increased sharply with a mean (±SD) C
max of 1.12 (±0.19) IU/mL. Thereafter, the decline of FVIII:C exhibited biphasic disposition characteristics. In the initial phase, the activity dropped at a rate consistent with relatively rapid but limited distribution into extravascular space. The mean (±SD) steady-state volume of distribution was 65.1 (± 35.1) mL/kg. During the terminal phase, the rate of decline in FVIII:C was slower than the initial phase with a mean (±SD) terminal elimination half-life of 11.8 (± 5.1) hours. A comparable pharmacokinetic profile was obtained after repeated use for six months. The ratios of geometric least square means of month 6-to-baseline pharmacokinetic were 107%, 100% and 104% for recovery, AUC
t and AUC
∞, respectively. No time-dependent changes in the pharmacokinetic properties of Xyntha were observed (Table 3). (See Table 3.)
Click on icon to see table/diagram/image
In a pivotal phase III study (Study 311) for surgical prophylaxis, Xyntha pharmacokinetics were evaluated during the perioperative management of patients with hemophilia A who were undergoing major surgery. At the baseline visit, all patients received a single dose of Xyntha of approximately 50 IU/kg. Plasma samples were analyzed for FVIII activity using a validated one-stage (OS) clotting method. Recovery data are available for a total of 30 patients; the mean (± standard deviation [SD]) K-value was 2.11(± 0.43) IU/dL per IU/kg, and the mean (±SD)
in vivo recovery value was 101.0% (± 20%).
Chromogenic Assay: The labeled potency of Xyntha manufactured by the previous process is based on the European Pharmacopoeia chromogenic substrate assay in which the Pfizer In-House Recombinant Factor VIII Potency Reference Standard has been calibrated to the WHO International Standard using the chromogenic substrate assay.
In a crossover pharmacokinetic study of eighteen (18) previously treated patients using the chromogenic assay, the circulating mean half-life for Xyntha manufactured by the previous process was 14.8 ± 5.6 hours (ranged from 7.6 - 28.5 hours), which was not statistically significantly different from plasma-derived Antihemophilic Factor (Human), (pdAHF), which had a mean half-life of 13.7 ± 3.7 hours (ranged from 8.8 - 25.1 hours). Mean incremental recovery (K-value) of Xyntha manufactured by the previous process in plasma was 2.4 ± 0.4 IU/dL per IU/kg (ranged from 1.9 - 3.3 IU/dL per IU/kg). This was comparable to the mean incremental recovery observed in plasma for pdAHF which was 2.3 ± 0.3 IU/dL per IU/kg (ranged from 1.7 - 2.9 IU/dL per IU/kg).
In additional clinical studies using Xyntha manufactured by the previous process, pharmacokinetic parameters measured using the chromogenic assay were determined for previously treated patients (PTPs) and previously untreated patients (PUPs). In PTPs (n=101; median age 26 ± 12 years), Xyntha manufactured by the previous process had a recovery at Week 0 of 2.4 ± 0.4 IU/dL per IU/kg (range 1.1 to 3.8 IU/dL per IU/kg). In measurements over 4 years of use (Month 3 [n=90], Month 6 [n=87], Month 12 [n=88], Month 24 [n=70], Month 36 [n=64] and Month 48 [n=52]), the mean incremental recovery was reproducible and ranged from 2.3 to 2.5 IU/dL per IU/kg. A subset of 37 study subjects had evaluable pharmacokinetic profiles at both baseline and Month 12. The 90% confidence intervals for the ratios of the mean values of Month 12-to-baseline AUC
T, AUC
∞, and K-value were well within the bioequivalence window of 80% to 125%, demonstrating the stability of these pharmacokinetic parameters over 1 year. In PUPs (n=59; median age 10 ± 8.3 months), Xyntha manufactured by the previous process had a mean recovery at Week 0 of 1.5 ± 0.6 IU/dL per IU/kg (range 0.2 to 2.8 IU/dL per IU/kg). The mean incremental recovery for PUPs was stable over time (5 visits during a 2-year period) and ranged from 1.5 to 1.8 IU/dL per IU/kg of Xyntha manufactured by the previous process. Population pharmacokinetic modeling using data from 44 PUPs led to a mean estimated half-life of Xyntha manufactured by the previous process in PUPs of 8.0 ± 2.2 hours. (See Table 4.)
Click on icon to see table/diagram/image
Special Populations and Conditions - Pediatrics: Table 5 shows the pharmacokinetic parameters of nine children, four aged 14 or 15 years of age, who are also included in the summary for the adults as previously mentioned, along with five children aged 3.7 to 5.8 years after Xyntha administration. Compared with adults, the half-life of factor VIII after Xyntha is shorter in children and clearance (based on body weight) is approximately 40% higher in children. (See Table 5.)
Click on icon to see table/diagram/image
Non-Clinical Toxicology: Single Dose Toxicity: BDDrFVIII was tested for potential toxicity of a single intravenous (IV) dose in Sprague-Dawley rats and cynomologus monkeys.
Sprague-Dawley rats (5/sex/group) were administered single doses of 0 (saline), 2500 or 10000 IU/kg of BDDrFVIII intravenously. Animals were observed for 14 days and sacrificed for gross pathology and histopathology. There were no adverse effects related to BDDrFVIII administration; therefore, the no-observed-adverse-effect-level (NoAEL) was ≥ 10000 IU/kg.
Two groups of cynomologus monkeys (1/sex/group) were administered alternate escalating single doses of 200, 400, 800, or 1600 IU/kg BDDrFVIII intravenously every third day in a dose-range finding toxicology study. The animals were subsequently allowed a 14-day washout period and used for a 7-day repeat dose study described as follows. As expected, there were transient dose-dependent increases in plasma factor VIII activity. During the single dose escalation study there were no adverse effects associated with BDDrFVIII administration; therefore, the NoAEL was ≥ 1600 IU/kg.
Repeated Dose Toxicity: BDDrFVIII was tested for potential toxicity of repeated IV doses in Sprague-Dawley rats and cynomologus monkeys.
Rat Studies: Sprague-Dawley rats (4/sex/group) were administered doses of 0 (vehicle), 200, 400, 800, or 1600 IU/kg/day BDDrFVIII intravenously once daily for 10 or 11 days. Satellite groups of 10 males each were included at dose levels of 200 and 800 IU/kg/day to monitor for antibodies against BDDrFVIII at 4 and 19 days after the last dose administration. There were significant BDDrFVIII antibody responses in all animals from the satellite treatment groups as expected when administering a human protein to animals. There were no adverse effects associated with BDDrFVIII administration; therefore, the NoAEL was ≥ 1600 IU/kg/day.
The general toxicity of BDDrFVIII was evaluated in a 4-week study in Sprague-Dawley rats. Animals (10/sex/group) were administered 0 (vehicle), 50, 250, or 1250 IU/kg/day BDDrFVIII intravenously once daily for 4 weeks. In addition to standard comprehensive assessments of clinical, clinical laboratory, and anatomic pathology, BDDrFVIII antibody responses were assessed at the end of the treatment period in a subset of the animals. There was a dose-related induction of antibodies reactive to BDDrFVIII in the treated animals. There were slight increases in APTT in a small number of animals from the 250 and 1250 IU/kg/day groups. These minor elevations in APTT were considered secondary to the anti-BDDrFVIII antibody responses which apparently have the potential for neutralization of exogenous human recombinant and endogenous rat factor VIII activity. This neutralization results in prolongation of APTT which is an ex vivo measure of the intrinsic and common pathways of coagulation. There were no adverse effects associated with BDDrFVIII administration; therefore, the NoAEL was ≥ 1250 IU/kg/day.
Cynomologus Monkey Studies: A 7-day repeat dose toxicity study in cynomologus monkeys was performed using the same animals as described in the single-dose escalation study with 4 additional naive animals. After a 14-day washout period, 0 (vehicle) (1/sex), 800 (2/sex), or 1250 (2/sex) IU/kg/day BDDrFVIII was administered intravenously once daily for 7 days. In addition to standard clinical laboratory and anatomic pathology assessments, animals were monitored for plasma factor VIII activity. Three of four previously treated animals had decreased plasma factor VIII activity on Day 7 compared to previously untreated animals suggesting the appearance of antibody responses resulting in neutralization of both exogenous recombinant human and endogenous factor VIII activity. Antibodies to BDDrFVIII were not measured in this study. There were no adverse effects associated with BDDrFVIII administration; therefore, the NoAEL was ≥ 1250 IU/kg/day.
The general toxicity of BDDrFVIII was evaluated in a 4-week study in cynomologus monkeys (3/sex/group) at doses of 0 (vehicle), 50, 250, or 1250 IU/kg/day BDDrFVIII intravenously. In addition to comprehensive clinical, clinical laboratory, and post-mortem examinations, plasma factor VIII activity was assessed prestudy and on Days 13, 20, and 28, and anti-BDDrFVIII antibodies and neutralizing activity were determined prestudy and on Day 28. One animal was found dead and three sacrificed when moribund. These deaths occurred on or shortly after days of scheduled bleeds (Days 20 and 28/29), and in each case the predominant signs were hemorrhage at sites of venipuncture and/or marked anemia. Clinical signs related to treatment were extensive hemorrhage and bruising at sites of venipuncture. Body weight losses were observed in two moribund animals, and reduced food intake was observed in one moribund animal. Dose and time-related appearance of anti-BDDrFVIII antibodies were associated with prolongation of APTT, decreases in red blood cell indices (PCV, Hb, and RBC count) and decreased plasma factor VIII activity in animals from the 250 and 1250 IU/kg/day groups.
Treatment-related gross lesions were limited to hemorrhage at sites of venipuncture and at scattered other locations. These changes were observed predominantly in animals from the 250 and 1250 IU/kg/day groups. Histologic changes related to treatment included hemorrhage at sites of venipuncture and in various other organs including the heart, subcutaneous tissues, urinary bladder, spinal canal, skeletal muscle, gastrointestinal tract, and connective tissues. The heart was apparently a predisposition site for histologic lesions of hemorrhage and edema with secondary inflammation and early fibrosis and, sometimes, associated regions of myocardial degeneration.
All adverse effects observed in this study were considered related to the neutralizing antibody response to both administered recombinant human and endogenous monkey factor VIII. Neutralizing antibody responses resulted in an acquired hemophilia syndrome and predisposition to multiorgan hemorrhage and the sequelae of death, moribundity, anemia, and hemorrhage-induced inflammation. The NoAEL for changes related to this syndrome was 50 IU/kg/day, but no adverse effects unrelated to the immunogenicity of BDDrFVIII were observed at any dose level.
After completing the 4-week IV toxicity study with BDDrFVIII in cynomologus monkeys, it was considered appropriate to conduct a similar study with a plasma-derived human factor VIII product, Octonativ-M, in order to demonstrate the comparability of the changes observed with BDDrFVIII to those with a plasma-derived factor VIII product. Cynomologus monkeys were administered daily doses of 0 (vehicle) (2/sex), 250 or 1250 (3/sex/group) IU/kg/day of Octonativ-M intravenously for 5 weeks. Parameters examined were similar to those in the BDDrFVIII 4-week study described previously.
One animal from the 250 IU/kg/day group was sacrificed moribund on Day 29. Clinical signs related to treatment were hemorrhage at sites of venipuncture. Anti-factor VIII antibodies were detected in all animals treated with Octonativ-M. Clinical laboratory changes included dose- and time-related increases in APTT, decreased red cell indices, increased fibrinogen, and decreased erythrocyte sedimentation rates. The longitudinal kinetics of onset of these changes correlated with dose- and time-related appearance of decreased plasma factor VIII activity and the appearance of plasma factor VIII inhibitors. Treatment-related post-mortem findings in animals from both treatment groups included gross hemorrhage at sites of venipuncture and in the heart. These changes correlated with microscopic lesions of hemorrhage and edema, with associated inflammation and early fibrosis and myocyte degeneration.
Overall, changes observed in this study with a plasma-derived human factor VIII product were analogous to those seen with BDDrFVIII. All adverse effects were considered secondary to the immunogenicity of the test molecule and the acquired hemophilic state of the animals. These effects occurred at IV doses of 250 and 1250 IU/kg/day. There was no NoAEL identified in this study of Octonativ-M.
A 6-week multidose subcutaneous (SC) and IV antigenicity study of BDDrFVIII, which included limited toxicology endpoints, was performed in cynomologus monkeys. In this study, animals were administered 0 (vehicle) (3/SC route only), 50 (6/route), or 250 IU/kg (4/route) BDDrFVIII either IV or SC every other day for 6 weeks. Limited clinical, hematologic, clinical chemistry, plasma factor VIII activity, factor VIII antibody, and plasma inhibitor determinations were performed. Both SC and IV administration resulted in antibody formation (2/6 animals in the low dose IV treatment group and 100% of the animals in all other groups). Inhibitor antibodies were detected in some animals after treatment by either route of administration (2/6 animals in the low dose IV-treated group, 4/4 animals in the high dose IV group, 1/6 animals in the low dose SC-treated group, and 2/4 animals in the high dose SC group). Gross necropsy and limited histopathology evaluations were performed. Two deaths were observed in the high dose IV administration group. These were considered secondary to hemorrhage and severe anemia. There were dose- and time-related increases in APTT and decreases in red blood cell indices by both routes. There were clinical and post-mortem observations of hemorrhage at venipuncture sites. Histologic changes of ischemic degeneration in the heart were seen in one animal from the 50 IU/kg IV group and one animal from the 250 IU/kg IV group. These changes were all considered related to the immunogenicity of BDDrFVIII in cynomologus monkeys, the acquired hemophilic syndrome, and resultant hemorrhagic anemia and myocardial ischemia. There was no NoAEL identified in this study.
Reproductive and Developmental Toxicology: No specific studies to investigate the potential effects of BDDrFVIII on reproductive or developmental function have been conducted. In the 4-week repeat dose toxicity studies, gonadal and secondary sex organs were examined by gross and histopathology, and there were no apparent effects of BDDrFVIII on these tissues.
Mutagenic Potential: BDDrFVIII was assessed for the potential to induce bone marrow micronuclei
in vivo in the CD-1 mouse. Animals (10/sex/group) were administered 0 (vehicle), 2490, 4980, or 9960 IU/kg/day BDDrFVIII IV for two consecutive days (Tabular Format and Kabi Pharmacia Report 9296824). Five animals/sex/group were sacrificed 24 and 48 hours after the second dose administration. It was concluded that BDDrFVIII did not induce micronuclei in the polychromatic erythrocytes of the bone marrow of mice at any dose level tested.
Repeat Dose: As part of the nonclinical evaluation of Xyntha, a 4-week toxicity study was conducted in cynomologus monkeys with Xyntha administered by intravenous (IV) injection at dosages of 0, 50, or 1250 IU/kg/day once daily for 29 or 30 days. The toxicity profile from this study was compared with that obtained in a previously conducted 4-week toxicity study in cynomologus monkeys with original BDDrFVIII administered by IV injection at dosages of 0, 50, 250, and 1250 IU/kg/day.
The administration of Xyntha to monkeys at an IV dosage of 1250 IU/kg/day for 29 or 30 days was generally well tolerated prior to the formation of anti-Xyntha antibodies. The immunologic response against Xyntha consisted of anti-Xyntha antibody production and increased FVIII inhibitors, which resulted in decreased FVIII activity that caused impairment of the coagulation pathway. These findings correlated with clinical pathology changes, hemorrhage, and tissue changes secondary to hemorrhage and were also observed in a previously conducted 4-week toxicity study in cynomologus monkeys with BDDrFVIII.
The most significant hematologic alteration was a dose-dependent increase in activated partial thromboplastin time (APTT) that was also observed in monkeys at 250 and 1250 IU/kg/day in the previous 4-week toxicity study with original BDDrFVIII. Absolute and relative (to body and brain) mean liver weights were increased (21% to 25%, compared to controls) in male monkeys given 1250 IU/kg/day. Individual liver weights were 74.9, 72.4, 57.6 g (absolute), 99.5, 102.0, 88.2 (% relative to brain weight) and 1.9, 2.1, 1.6 (% relative to body weight) in control males and 80.8, 79.6, 88.5 g (absolute), 123.2, 121.9, 116.8 (% relative to brain weight) and 2.4, 2.3, 2.3 (% relative to body weight) in males given 1250 IU/kg/day. The weight increase was not considered to be toxicologically significant due to the low magnitude of change and the lack of macroscopic or microscopic correlates.
Local Tolerance Studies: A separate local tolerance study was not conducted with Xyntha. Instead, local injection sites were evaluated macroscopically and microscopically in the repeat-dose study conducted in monkeys. Macroscopically, red discoloration occurred at the injection sites of all monkeys regardless of treatment (including controls). Microscopically, slight to moderate perivascular and vascular neutrophilic inflammation and/or fibrosis often accompanied hemorrhage at the injection sites in animals given Xyntha, as well as in controls. Hemorrhage was more severe in monkeys given Xyntha (slight to marked at 50 IU/kg/day and mild to severe at 1250 IU/kg/day) when compared with controls (mild to moderate). The macroscopic and microscopic observations with Xyntha were similar to findings in the previous 4-week toxicity study in cynomologus monkeys conducted with original BDDrFVIII.
Other Toxicity Studies: Because a novel affinity ligand (TN8.2) is used in the purification process for Xyntha, a non-GLP-compliant, single-dose toxicity study was conducted in rats to assess the acute toxicity of TN8.2 in the event that the peptide leached from the chromatographic resin into the product stream during purification. Because TN8.2 binds to FVIII,
in vitro studies were also conducted to evaluate the possible effects of TN8.2 on the clotting activity of rat plasma, human plasma, or Xyntha. TN8.2 has not been detected in any batch of drug substance tested to date.
Single-dose administration of the TN8.2 affinity ligand in rats at 0.6 mg/kg was well tolerated and did not induce acute toxicity.
Microbiology: No microbiological information is required for this drug product.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image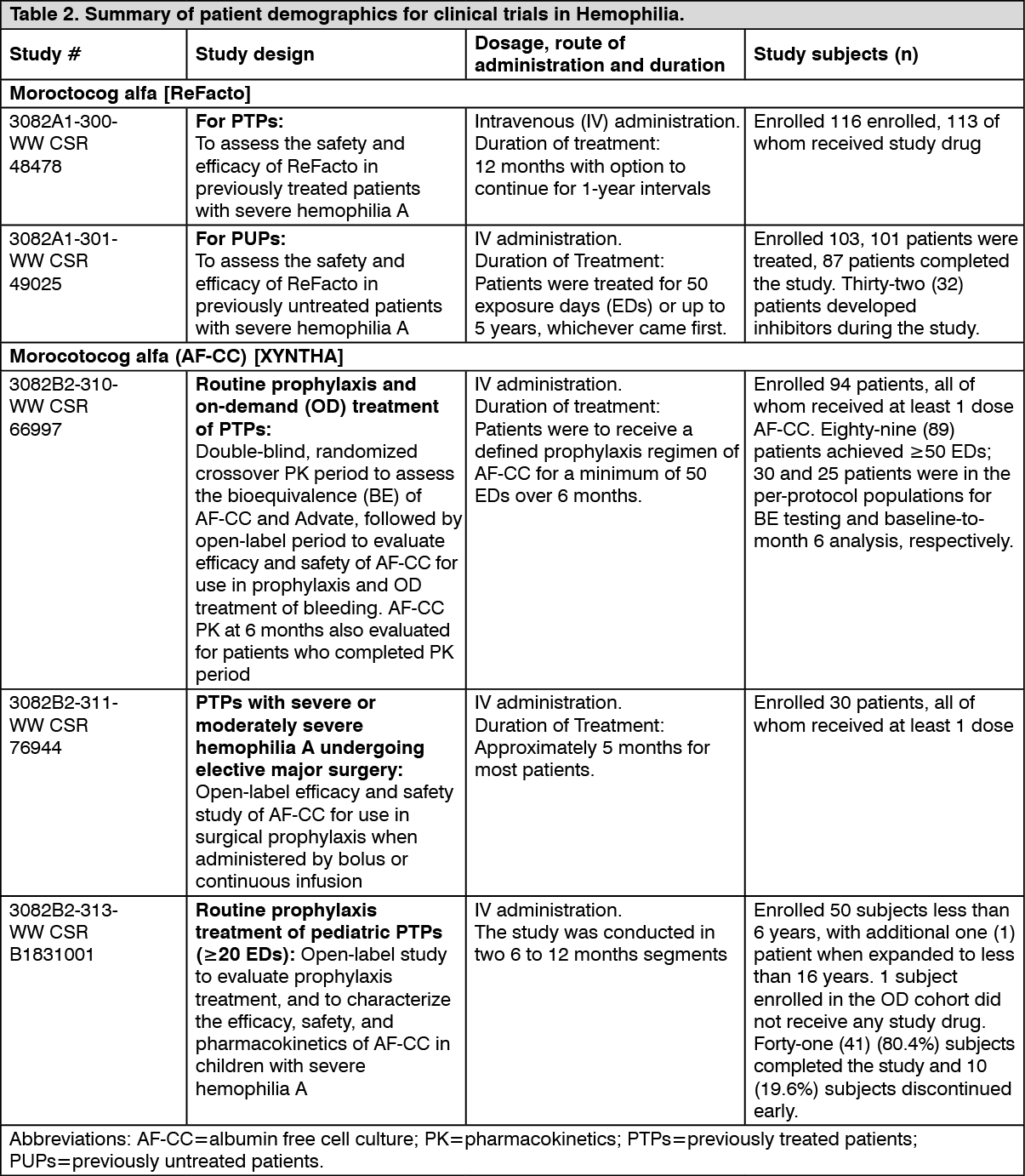 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image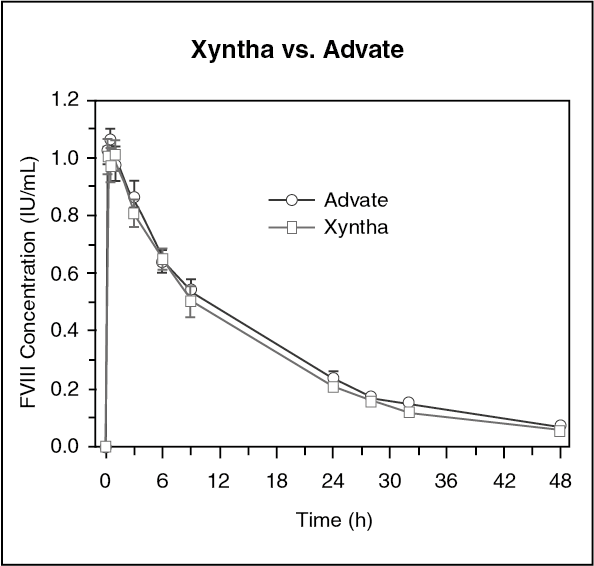 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image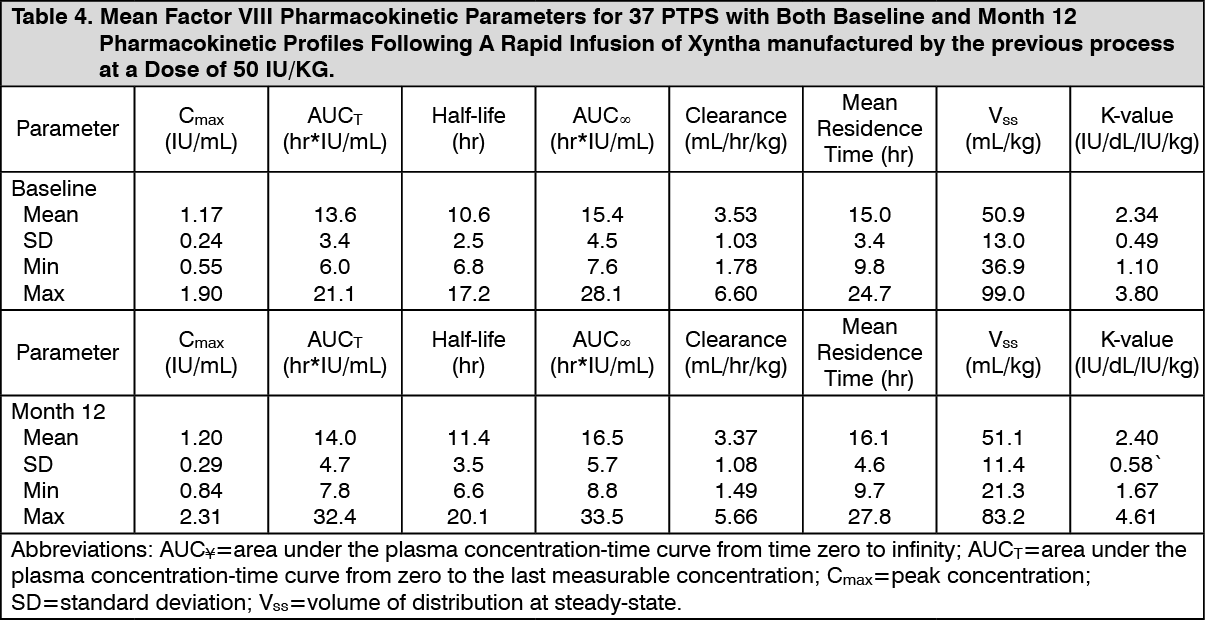 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image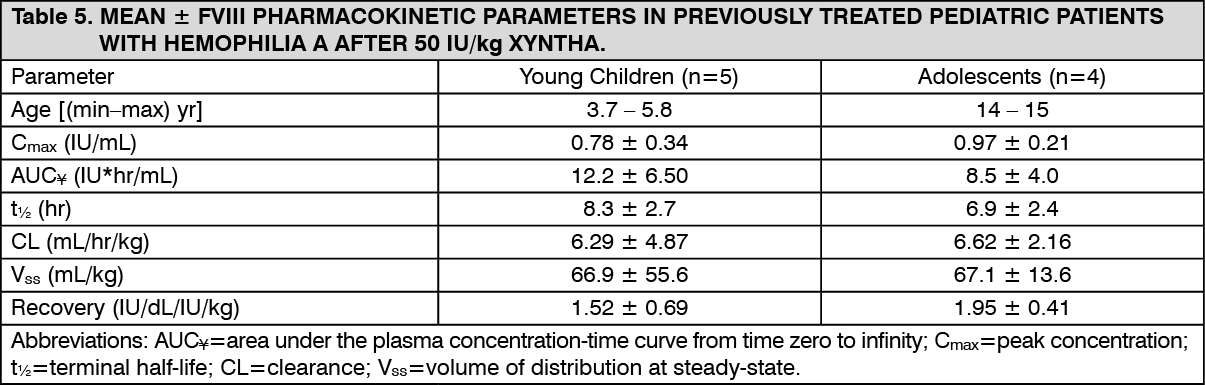 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image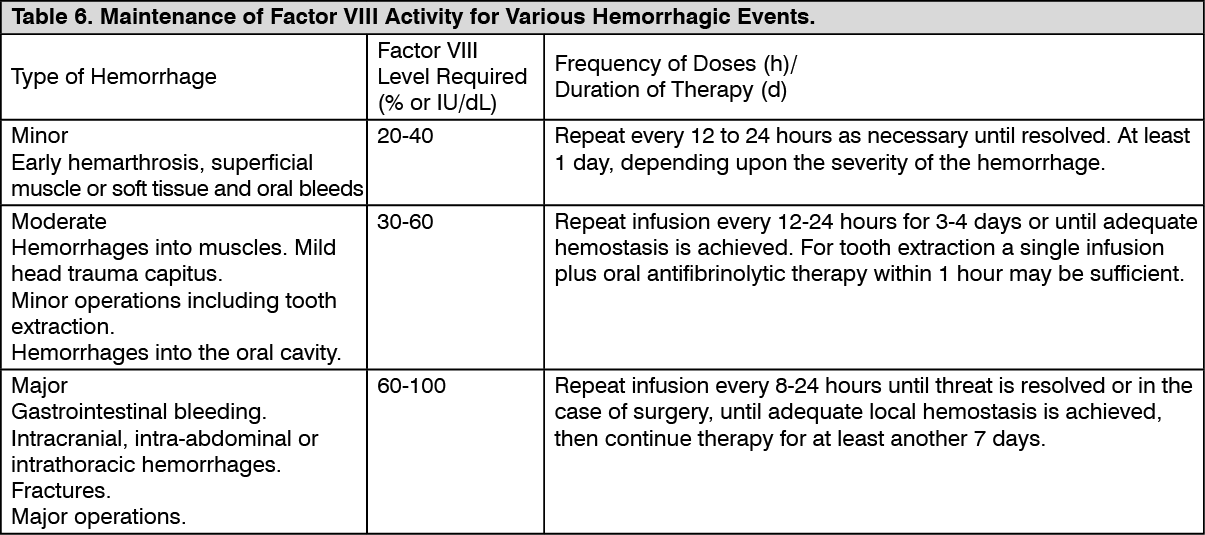 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image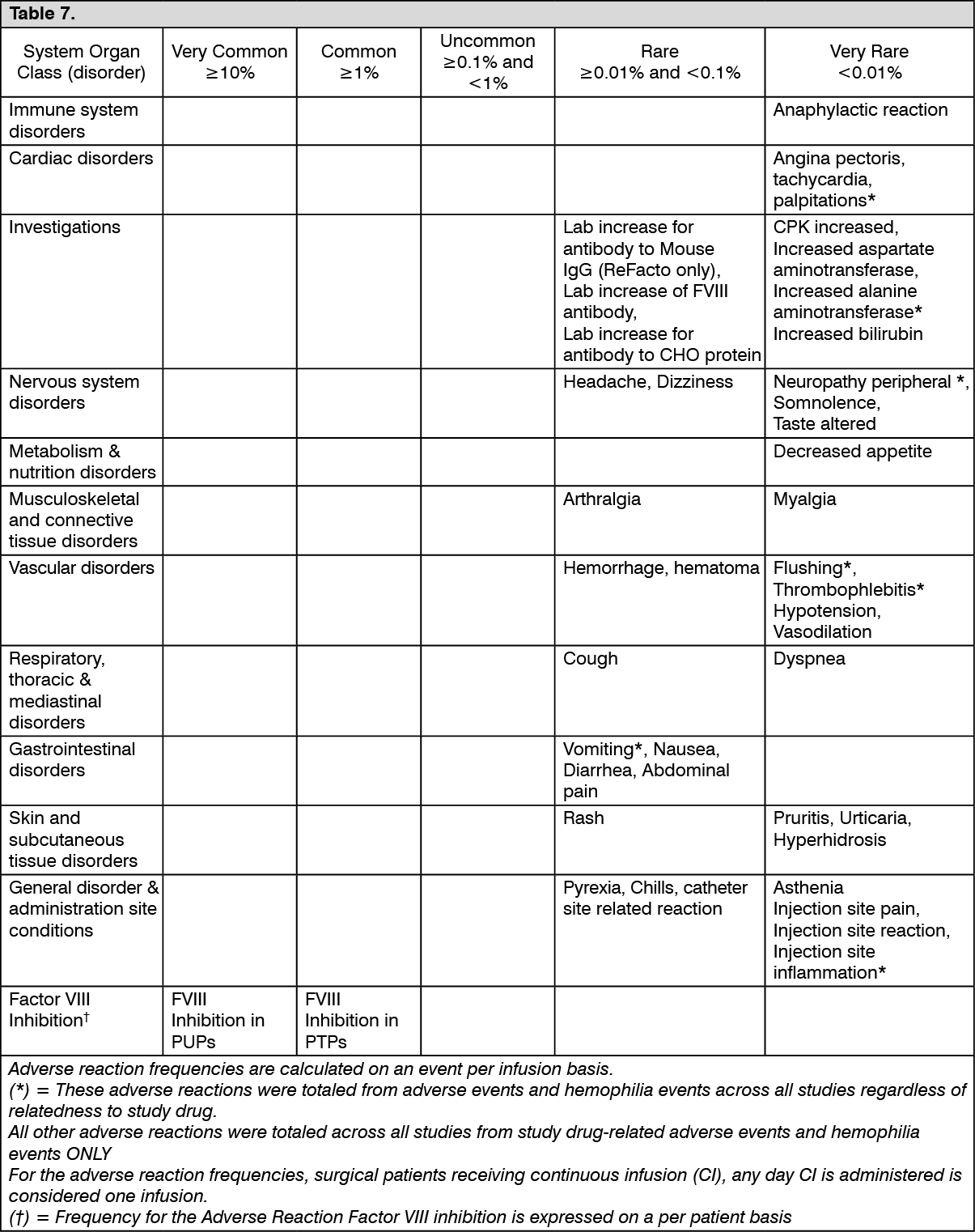 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
