Pharmacotherapeutic group: antihaemorrhagics, blood coagulation factor VIII.
ATC code: B02BD02.
PHARMACOLOGY: Pharmacodynamics: The factor VIII/von Willebrand factor complex consists of two molecules (factor VIII and von Willebrand factor) with different physiological functions. When infused into a haemophilic patient, factor VIII binds to von Willebrand factor in the patient's circulation. Activated factor VIII acts as a cofactor for activated factor IX, accelerating the conversion of factor X to activated factor X. Activated factor X converts prothrombin into thrombin. Thrombin then converts fibrinogen into fibrin and a clot can be formed. Haemophilia A is a X-chromosomal linked hereditary disorder of blood coagulation due to decreased levels of factor VIII:C and results in profuse bleeding into joints, muscles or internal organs, either spontaneously or as results of accidental or surgical trauma. By replacement therapy the plasma levels of factor VIII are increased, thereby enabling a temporary correction of the factor deficiency and correction of the bleeding tendencies.
Rurioctocog alfa pegol, is a pegylated recombinant human factor VIII with an extended half-life. Rurioctocog alfa pegol is a covalent conjugate of octocog alfa consisting of 2,332 amino acids with polyethylene glycol (PEG) reagent (MW 20 kDa). The therapeutic activity of rurioctocog alfa pegol is derived from octocog alfa, which is produced by recombinant DNA technology from a Chinese hamster ovary cell line. Octocog alfa is then covalently conjugated with the PEG reagent. The PEG moiety is conjugated to octocog alfa to increase the plasma half-life.
Clinical efficacy and safety: The safety, efficacy, and pharmacokinetics of ADYNOVATE were evaluated in a pivotal multicenter, open-label, prospective clinical trial that compared the efficacy of a twice weekly prophylactic treatment regimen to on-demand treatment and determined haemostatic efficacy in the treatment of bleeding episodes. A total of 137 male PTPs (12 to 65 years of age) with severe haemophilia A received at least one infusion with ADYNOVATE. Twenty-five of the 137 subjects were adolescents (12 to less than 18 years of age).
Prophylactic treatment: Subjects received either prophylactic treatment (n = 120) with ADYNOVATE at a dose of 40-50 IU per kg twice weekly or on-demand treatment (n = 17) with ADYNOVATE at a dose of 10-60 IU per kg for a 6-month period. The median dosing interval was 3.6 days and the mean dose (SD) was 48.7 (4.4) IU/kg. One hundred eighteen of 120 (98%) prophylaxis subjects remained on the starting recommended regimen without dose adjustment, and 2 subjects increased their dose to 60 IU/kg during prophylaxis due to bleeding in target joints.
In the per-protocol population, i.e. dosed according to the protocol specific dosing requirements, a total of 101 subjects received a twice a week regimen in the prophylaxis arm, and 17 subjects were treated episodically in the on-demand arm. The median annualised bleed rate (ABR) in the on-demand treatment arm was 41.5 compared to 1.9 while on a twice a week prophylaxis regimen. The median joint ABR (Q1 ; Q3) in the on-demand arm was 38.1 (24.5 ; 44.6) compared to 0.0 (0.0 ; 2.0) while on prophylaxis, and the median spontaneous ABR was 21.6 (11.2 ; 33.2) on the on-demand arm compared to 0.0 (0.0 ; 2.2) while on prophylaxis. Results for the full-analysis population were similar to those for the per-protocol population. Of note, ABR is not comparable between different factor concentrates and between different clinical studies.
Forty out of 101 subjects (40%) experienced no bleeding episodes, 58 out of 101 subjects (57%) experienced no joint bleeding episodes, and 58 out of 101 subjects (57%) experienced no spontaneous bleeding episodes in the prophylaxis arm. All subjects in the on-demand arm experienced a bleeding episode, including a joint or spontaneous bleeding episode.
Treatment of bleeding episodes: A total of 518 bleeding episodes were treated with ADYNOVATE in the per-protocol population. Of these, 361 bleeding episodes (n=17 subjects) occurred in the on-demand arm and 157 (n=61 subjects) occurred in the prophylaxis arm. The median dose per infusion to treat all bleeding episodes in the per-protocol population was 32.0 (Interquartile Range (IQR): 21.5) IU per kg. Overall, 95.9% of bleeding episodes were controlled with 1 to 2 infusions and 85.5% were controlled with only 1 infusion. Of the 518 bleeding episodes, 96.1% were rated excellent (full relief of pain and cessation of objective signs of bleeding after a single infusion) or good (definite pain relief and/or improvement in signs of bleeding after a single infusion) in their response to treatment with ADYNOVATE.
Paediatric population < 12 years of age: A total of 66 PTPs with severe haemophilia A were dosed (32 subjects aged < 6 years and 34 subjects aged 6 to < 12 years) in the paediatric study. The prophylactic regimen was 40 to 60 IU/kg of ADYNOVATE twice a week. The mean dose (SD) was 54.3 (6.3) IU/kg and the median frequency of infusions per week was 1.87. The median overall ABR was 2.0 (IQR: 3.9) for the 65 subjects in the per-protocol population and the median ABRs for spontaneous and joint bleeding episodes were both 0 (IQR: 1.9). Twenty four out of 65 subjects (37%) experienced no bleeding episodes, 47 out of 65 subjects (72%) experienced no joint bleeding episodes, and 43 out of 65 subjects (66%) experienced no spontaneous bleeding episodes on prophylaxis.
Of the 70 bleeding episodes observed during the paediatric study, 82.9% were controlled with 1 infusion and 91.4% were controlled with 1 or 2 infusions. Control of bleeding was rated excellent (full relief of pain and cessation of objective signs of bleeding after a single infusion) or good (definite pain relief and/or improvement in signs of bleeding after a single infusion) in 63 out of 70 (90.0%) bleeding episodes.
Perioperative management (surgical prophylaxis): A total of 21 major surgical procedures and 5 additional minor surgeries were performed and assessed in 21 unique subjects in the surgery study. For major surgeries, the preoperative loading dose ranged from 36 IU/kg to 109 IU/kg (median: 63 IU/kg); and postoperative total dose ranged from 186 IU/kg to 1320 IU/kg (median: 490 IU/kg). The median total dose for major surgeries was 553 IU/kg (range: 248-1394 IU/kg) and the median total dose of minor surgeries was 106 IU/kg (range: 76-132 IU/kg).
Perioperative haemostatic efficacy was rated as excellent (blood loss less than or equal to that expected for the same type of procedure performed in a non-haemophilic patient, and required blood components for transfusions less than or similar to that expected in non-haemophilic population) for all 26 (21 major, 5 minor) procedures. The median (IQR) observed intraoperative blood loss (n = 14) was 10.0 (20.0) ml compared to the predicted average blood loss (n = 14) of 150.0 (140.0) ml for major orthopaedic surgeries.
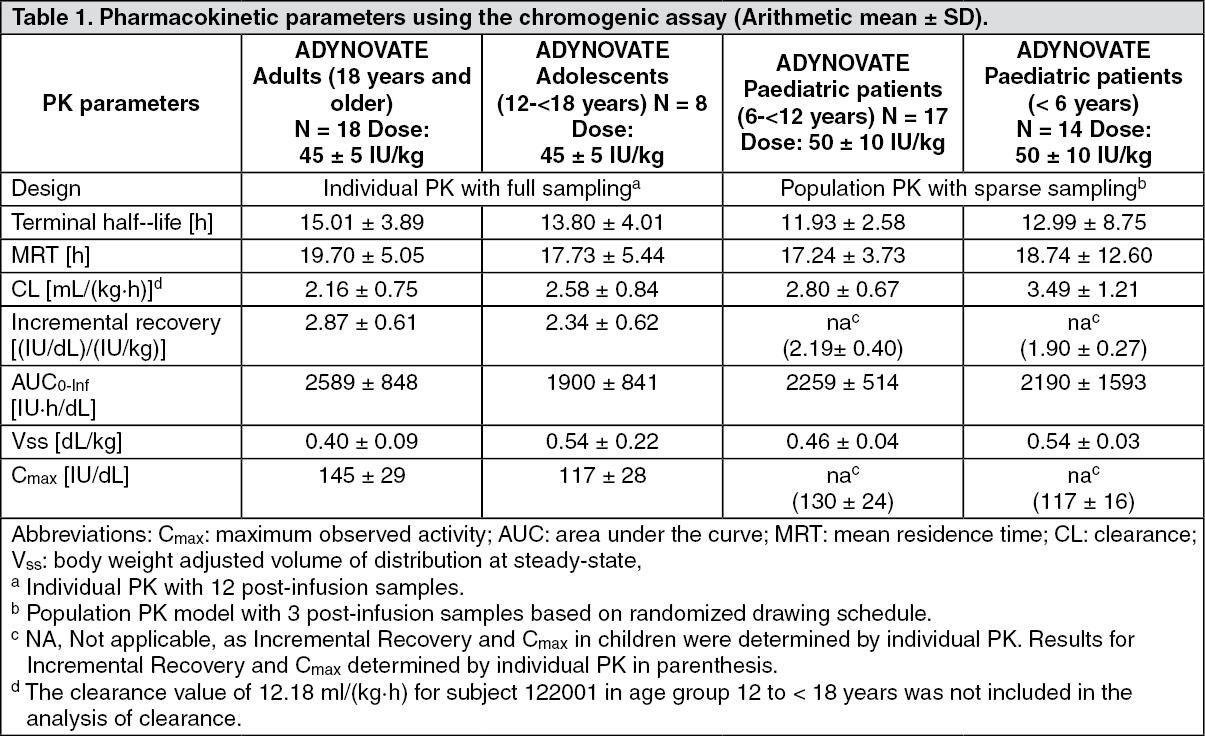
Pharmacokinetics: The pharmacokinetics (PK) of ADYNOVATE were evaluated in a crossover study with octocog alfa in 26 subjects (18 adults and 8 adolescents) and in 22 subjects (16 adults and 6 adolescents) after 6 months of treatment with ADYNOVATE. Plasma factor VIII activity was measured by the one stage clotting assay and chromogenic assay.
ADYNOVATE has an extended half-life of 1.4 to 1.5-fold compared to recombinant human coagulation factor VIII (octocog alfa) in the adolescent and adult population, as determined based on one stage clotting and chromogenic assays, respectively. An increase in AUC and a decrease in clearance as compared to the parent molecule, octocog alfa, were also observed. Incremental recovery was comparable with both products. The change in PK parameters was similar in both the adult and adolescent populations and between one-stage clotting and chromogenic substrate assays.
Paediatric Pharmacokinetics: Pharmacokinetic parameters calculated from 39 subjects less than 18 years of age (intent-to-treat analysis) are available for 14 children (2 to less than 6 years), 17 older children (6 to less than 12 years) and 8 adolescent subjects (12 to < 18 years of age). The half-life extension in the paediatric population was 1.3 to 1.5 fold using both the one stage clotting and chromogenic assays. The mean clearance (based on body weight) of ADYNOVATE was higher and the mean half-life was lower in children less than 12 years of age than adults.
A higher dose may be required in children less than 12 years of age, see Dosage & Administration. (See Table 1.)
 Click on icon to see table/diagram/image
Toxicology: Preclinical safety data:
Click on icon to see table/diagram/image
Toxicology: Preclinical safety data: In the repeat dose toxicity study in Cynomologous monkey, two animals showed vacuolation in the kidney in the mid dose group (350IU/kg). The vacuolations did not recover after 2 weeks. The human relevance of kidney vacuolation observed in the preclinical study is unknown.
Nonclinical data are limited to 1 month exposure and no studies in juvenile animals were conducted with ADYNOVATE. Thus it was not possible to conclude on the potential risks of PEG accumulation in various tissues/organs relevant for chronic use of ADYNOVATE in the paediatric population. No studies on genotoxicity, carcinogenicity or reproductive toxicity have been performed with ADYNOVATE.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
