Pharmacotherapeutic group: Antineoplastic agents, protein kinase inhibitors.
ATC code: L01EX01.
PHARMACOLOGY: Pharmacodynamics: Mechanism of action: Sunitinib inhibits multiple RTKs that are implicated in tumour growth, neoangiogenesis, and metastatic progression of cancer. Sunitinib was identified as an inhibitor of platelet-derived growth factor receptors (PDGFRα and PDGFRβ), vascular endothelial growth factor receptors (VEGFR1, VEGFR2, and VEGFR3), stem cell factor receptor (KIT), Fms-like tyrosine kinase-3 (FLT3), colony stimulating factor receptor (CSF-1R), and the glial cell-line derived neurotrophic factor receptor (RET). The primary metabolite exhibits similar potency compared to sunitinib in biochemical and cellular assays.
Clinical efficacy and safety: The clinical safety and efficacy of sunitinib has been studied in the treatment of patients with GIST who were resistant to imatinib (i.e., those who experienced disease progression during or following treatment with imatinib) or intolerant to imatinib (i.e., those who experienced significant toxicity during treatment with imatinib that precluded further treatment), the treatment of patients with MRCC, and the treatment of patients with unresectable pNET.
Efficacy is based on time-to-tumour progression (TTP) and an increase in survival in GIST, on progression-free survival (PFS) and objective response rates (ORR) for treatment-naïve and cytokine-refractory MRCC respectively, and on PFS for pNET.
Gastrointestinal stromal tumours: An initial open-label, dose-escalation study was conducted in patients with GIST after failure of imatinib (median maximum daily dose 800 mg) due to resistance or intolerance. Ninety-seven patients were enrolled at various doses and schedules; 55 patients received 50 mg at the recommended treatment Schedule 4 weeks on/2 weeks off ("Schedule 4/2").
In this study, the median TTP was 34.0 weeks (95% CI: 22.0, 46.0).
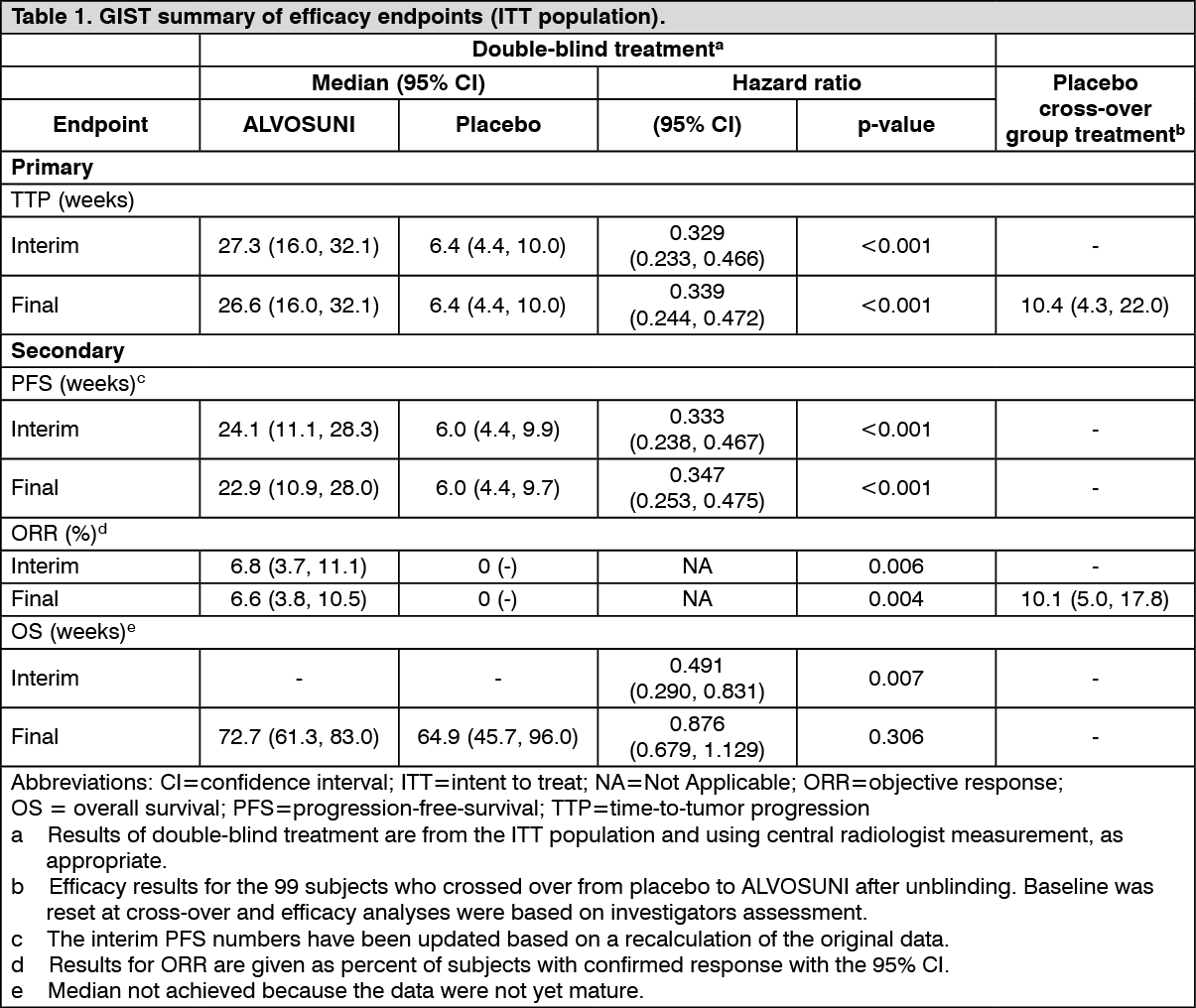
A Phase 3, randomised, double-blind, placebo-controlled study of sunitinib was conducted in patients with GIST who were intolerant to, or had experienced disease progression during or following treatment with imatinib (median maximum daily dose 800 mg). In this study, 312 patients were randomised (2:1) to receive either 50 mg sunitinib or placebo, orally once daily on Schedule 4/2 until disease progression or withdrawal from the study for another reason (207 patients received sunitinib and 105 patients received placebo). The primary efficacy endpoint of the study was TTP, defined as the time from randomisation to first documentation of objective tumour progression. At the time of the prespecified interim analysis, the median TTP on sunitinib was 28.9 weeks (95% CI: 21.3, 34.1) as assessed by the investigator and 27.3 weeks (95% CI: 16.0, 32.1) as assessed by the independent review and was statistically significantly longer than the TTP on placebo of 5.1 weeks (95% CI: 4.4, 10.1) as assessed by the investigator and 6.4 weeks (95% CI: 4.4, 10.0) as assessed by the independent review. The difference in overall survival (OS) was statistically in favour of sunitinib [hazard ratio (HR): 0.491; (95% CI: 0.290, 0.831)]; the risk of death was 2 times higher in patients in the placebo arm compared to the sunitinib arm.
After the interim analysis of efficacy and safety, at the recommendation of the independent Data and Safety Monitoring Board (DSMB), the study was unblinded and patients on the placebo arm were offered open-label sunitinib treatment.
A total of 255 patients received sunitinib in the open-label treatment phase of the study, including 99 patients who were initially treated with placebo.
The analyses of primary and secondary endpoints in the open-label phase of the study reaffirmed the results obtained at the time of the interim analysis, as shown in Table 1: See Table 1.
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Median OS in the ITT population was 72.7 weeks and 64.9 weeks (HR: 0.876; 95% CI: 0.679, 1.129; p=0.306), in the sunitinib and placebo arms, respectively. In this analysis, the placebo arm included those patients randomised to placebo who subsequently received open-label sunitinib treatment.
Treatment-naïve metastatic renal cell carcinoma: A Phase 3, randomised, multi-centre, international study evaluating the efficacy and safety of sunitinib compared with IFN-α in treatment-naïve MRCC patients was conducted. Seven hundred and fifty patients were randomised 1:1 to the treatment arms; they received treatment with either sunitinib in repeated 6-week cycles, consisting of 4 weeks of 50 mg daily oral administration followed by 2 weeks of rest (Schedule 4/2), or IFN-α, administered as a subcutaneous injection of 3 million units (MU) the first week, 6 MU the second week, and 9 MU the third week and thereafter, on 3 nonconsecutive days each week. The median duration of treatment was 11.1 months (range: 0.4-46.1) for sunitinib treatment and 4.1 months (range: 0.1-45.6) for IFN-α treatment. Treatment-related serious adverse events (TRSAEs) were reported in 23.7% of patients receiving sunitinib and in 6.9% of patients receiving IFN-α. However, the discontinuation rates due to adverse events were 20% for sunitinib and 23% for IFN-α. Dose interruptions occurred in 202 patients (54%) on sunitinib and 141 patients (39%) on IFN-α. Dose reductions occurred in 194 patients (52%)on sunitinib and 98 patients (27%) on IFN-α. Patients were treated until disease progression or withdrawal from the study. The primary efficacy endpoint was PFS. A planned interim analysis showed a statistically significant advantage for sunitinib over IFN-α, in this study, the median PFS for the sunitinib-treated group was 47.3 weeks, compared with 22.0 weeks for the IFN-α-treated group; the HR was 0.415 (95% CI: 0.320, 0.539; p-value <0.001). Other endpoints included ORR, OS, and safety. Core radiology assessment was discontinued after the primary endpoint had been met. At the final analysis, the ORR as determined by the investigator's assessment was 46% (95% CI: 41%, 51%) for the sunitinib arm and 12.0%(95% CI: 9%, 16%) for the IFN-α arm (p<0.001).
Sunitinib treatment was associated with longer survival compared to IFN-α. The median OS was 114.6 weeks for the sunitinib arm (95% CI: 100.1, 142.9) and 94.9 weeks for the IFN-α arm (95% CI: 77.7, 117.0) with a hazard ratio of 0.821 (95% CI: 0.673, 1.001; p=0.0510 by unstratified log rank).
The overall PFS and OS, observed in the ITT population, as determined by the core radiology laboratory assessment, are summarised in Table 2. (See Table 2.)
Click on icon to see table/diagram/image
Cytokine-refractory metastatic renal cell carcinoma: A Phase 2 study of sunitinib was conducted in patients who were refractory to prior cytokine therapy with interleukin-2 or IFN-α. Sixty-three patients received a starting dose of 50 mg sunitinib orally, once daily for 4 consecutive weeks followed by a 2-week rest period, to comprise a complete cycle of 6 weeks (Schedule 4/2). The primary efficacy endpoint was ORR, based on Response Evaluation Criteria in Solid Tumours (RECIST).
In this study, the objective response rate was 36.5% (95% CI: 24.7%, 49.6%) and the median TTP was 37.7 weeks (95% CI: 24.0, 46.4).
A confirmatory, open-label, single-arm, multi-centre study evaluating the efficacy and safety of sunitinib was conducted in patients with MRCC who were refractory to prior cytokine therapy. One hundred and 6 patients received at least one 50 mg dose of sunitinib on Schedule 4/2.
The primary efficacy endpoint of this study was ORR. Secondary endpoints included TTP, duration of response (DR) and OS.
In this study, the ORR was 35.8% (95% CI: 26.8%, 47.5%). The median DR and OS had not yet been reached.
Pancreatic neuroendocrine tumours: A supportive Phase 2, open-label, multi-centre study evaluated the efficacy and safety of single-agent sunitinib 50 mg daily on Schedule 4/2 in patients with unresectable pNET. In a pancreatic islet cell tumour cohort of 66 patients, the primary endpoint of response rate was 17%.
A pivotal Phase 3, multi-centre, international, randomised, double-blind, placebo-controlled study of single-agent sunitinib was conducted in patients with unresectable pNET.
Patients were required to have documented progression, based on RECIST, within the prior 12 months and were randomised (1:1) to receive either 37.5 mg sunitinib once daily without a scheduled rest period (N=86) or placebo (N=85).
The primary objective was to compare PFS in patients receiving sunitinib versus patients receiving placebo. Other endpoints included OS, ORR, PROs, and safety.
Demographics were comparable between the sunitinib and placebo groups. Additionally, 49% of sunitinib patients had nonfunctioning tumours versus 52% of placebo patients and 92% of patients in both arms had liver metastases.
Use of somatostatin analogues was allowed in the study.
A total of 66% of sunitinib patients received prior systemic therapy compared with 72% of placebo patients. In addition, 24% of sunitinib patients had received somatostatin analogues compared with 22% of placebo patients.
A clinically significant advantage in investigator-assessed PFS for sunitinib over placebo was observed. The median PFS was 11.4 months for the sunitinib arm compared to 5.5 months for the placebo arm [hazard ratio: 0.418 (95% CI: 0.263, 0.662), p-value=0.0001]; similar results were observed when derived tumour response assessments based upon application of RECIST to investigator tumour measurements were used to determine disease progression, as shown in Table 3. A hazard ratio favouring sunitinib was observed in all subgroups of baseline characteristics evaluated, including an analysis by number of prior systemic therapies. A total of 29 patients in the sunitinib arm and 24 in the placebo arm had received no prior systemic treatment; among these patients, the hazard ratio for PFS was 0.365 (95% CI: 0.156, 0.857), p=0.0156. Similarly, among 57 patients in the sunitinib arm (including 28 with 1 prior systemic therapy and 29 with 2 or more prior systemic therapies) and 61 patients in the placebo arm (including 25 with 1 prior systemic therapy and 36 with 2 or more prior systemic therapies), the hazard ratio for PFS was 0.456 (95% CI: 0.264, 0.787), p=0.0036.
A sensitivity analysis of PFS was conducted where progression was based upon investigator-reported tumour measurements and where all subjects censored for reasons other than study termination were treated as PFS events. This analysis provided a conservative estimate of the treatment effect of sunitinib and supported the primary analysis, demonstrating a hazard ratio of 0.507 (95% CI: 0.350, 0.733), p=0.000193. The pivotal study in pancreatic NET was terminated prematurely at the recommendation of an independent drug monitoring committee and the primary endpoint was based upon investigator assessment, both of which may have affected the estimates of the treatment effect.
In order to rule out bias in the investigator-based assessment of PFS, a BICR of scans was performed; this review supported the investigator assessment, as shown in Table 3. (See Table 3 and figure.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
OS data were not mature at the time of the study closure [20.6 months (95% CI: 20.6, NR) for the sunitinib arm compared to NR (95% CI: 15.5, NR) for the placebo arm, hazard ratio: 0.409 (95% CI: 0.187, 0.894), p-value=0.0204]. There were 9 deaths in the sunitinib arm and 21 deaths in the placebo arm.
Upon disease progression, patients were unblinded and placebo patients were offered access to open-label sunitinib in a separate extension study. As a result of the early study closure, remaining patients were unblinded and offered access to open-label sunitinib in an extension study. A total of 59 out of 85 patients (69.4%) from the placebo arm crossed over to open-label sunitinib following disease progression or unblinding at study closure. OS observed after 5 years of follow-up in the extension study showed a hazard ratio of 0.730 (95% CI: 0.504, 1.057).
Results from the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire (EORTC QLQ-C30) showed that the overall global health-related quality of life and the 5 functioning domains (physical, role, cognitive, emotional, and social) were maintained for patients on sunitinib treatment as compared to placebo with limited adverse symptomatic effects.
A Phase 4 multinational, multi-centre, single-arm, open-label study evaluating the efficacy and safety of sunitinib was conducted in patients with progressive, advanced/metastatic, well-differentiated, unresectable pNET.
One hundred six patients (61 patients in the treatment-naïve cohort and 45 patients in the later-line cohort) received treatment with sunitinib orally at 37.5 mg once a day on a continuous daily dosing (CDD) schedule.
The investigator-assessed median PFS was 13.2 months, both in the overall population (95% CI: 10.9, 16.7) and in the treatment-naïve cohort (95% CI: 7.4, 16.8).
Paediatric population: Experience on the use of sunitinib in paediatric patients is limited (see Dosage & Administration).
A Phase 1 dose-escalation study of oral sunitinib was conducted in 35 patients comprised of 30 paediatric patients (aged 3 years to 17 years) and 5 young adult patients (aged: 18 years to 21 years), with refractory solid tumours, the majority of whom were enrolled with a primary diagnosis of brain tumour. Dose-limiting cardiotoxicity was observed in the first part of the study which was therefore amended to exclude patients with previous exposure to potentially cardiotoxic therapies (including anthracyclines) or cardiac radiation. In the second part of the study, including patients with prior anticancer therapy but without risk factors for cardiac toxicity, sunitinib was generally tolerable and clinically manageable at the dose of 15 mg/m
2 daily (MTD) on Schedule 4/2. None of the subjects achieved complete response or partial response. Stable disease was observed in 6 patients (17%). One patient with GIST was enrolled at the 15 mg/m
2 dose level with no evidence of benefit. The observed adverse drug reactions were similar overall to those seen in adults (see Adverse Reactions).
A Phase 2 open-label study was conducted in 29 patients comprised of 27 paediatric patients (aged 3 years to 16 years) and 2 young adult patients (aged 18 years to 19 years) with HGG or ependymoma. The study was closed at the time of planned interim analysis due to the lack of disease control. Median PFS was 2.3 months in the HGG group and 2.7 months in the ependymoma group. Median overall OS was 5.1 months in the HGG group and 12.3 months in the ependymoma group. The most common (≥10%) reported treatment-related adverse events in patients in both groups combined were neutrophil count decreased (6 patients [20.7%]) and haemorrhage intracranial (3 patients [10.3%]) (see Adverse Reactions).
Evidence from a Phase
1/
2 study of oral sunitinib conducted in 6 paediatric patients with GIST aged 13 years to 16 years who received sunitinib on Schedule 4/2, at doses ranging between 15 mg/m
2 daily and 30 mg/m
2 daily, and available published data (20 paediatric or young adult patients with GIST) indicated that sunitinib treatment resulted in disease stabilization in 18 of 26 (69.2%) patients, either after imatinib failure or intolerance (16 patients with stable disease out of 21), or de novo/after surgery (2 patients with stable disease out of 5). In the Phase
1/
2 study, stable disease and disease progression was observed in 3 out of 6 patients each (1 patient received neo adjuvant and 1 patient received adjuvant imatinib, respectively). In the same study, 4 out of 6 patients (66.7%) experienced Grade 3-4 treatment-related adverse events (Grade 3 hypophosphataemia, neutropenia, and thrombocytopenia in 1 patient each and a Grade 4 neutropenia in 1 patient). In addition, the publications reported the following Grade 3 adverse drug reactions experienced by 5 patients: fatigue (2), gastrointestinal adverse drug reactions (including diarrhoea) (2), haematologic adverse drug reactions (including anaemia) (2), cholecystitis (1), hyperthyroidism (1), and mucositis (1).
A population pharmacokinetic (PK) and pharmacokinetic/pharmacodynamic (PK/PD) analysis was conducted with the scope to extrapolate the PK and key safety and efficacy endpoints of sunitinib in paediatric patients with GIST (aged: 6 years to 17 years). This analysis was based on data collected from adults with GIST or solid tumours and from paediatric patients with solid tumours. Based on the modelling analyses, the younger age and lower body size did not appear to affect negatively the safety and efficacy responses to sunitinib plasma exposures. Sunitinib benefit/risk did not appear to be negatively affected by younger age or lower body size, and was mainly driven by its plasma exposure.
The EMA has waived the obligation to submit the results of studies with sunitinib in all subsets of the paediatric population for the treatment of kidney or renal pelvis carcinoma (excluding nephroblastoma, nephroblastomatosis, clear cell sarcoma, mesoblastic nephroma, renal medullary carcinoma, and rhabdoid tumour of the kidney) (see Dosage & Administration).
The EMA has waived the obligation to submit the results of the studies with sunitinib in all subsets of the paediatric population for the treatment of gastroenteropancreatic neuroendocrine tumours (excluding neuroblastoma, neuroganglioblastoma, and phaeochromocytoma) (see Dosage & Administration).
Pharmacokinetics: The PK of sunitinib were evaluated in 135 healthy volunteers and 266 patients with solid tumours. The PK were similar in all solid tumours populations tested and in healthy volunteers.
In the dosing ranges of 25 to 100 mg, the area under the plasma concentration-time curve (AUC) and Cmax increase proportionally with dose. With repeated daily administration, sunitinib accumulates 3- to 4-fold and its primary active metabolite accumulates 7- to 10-fold. Steady-state concentrations of sunitinib and its primary active metabolite are achieved within 10 to 14 days. By Day 14, combined plasma concentrations of sunitinib and its active metabolite are 62.9-101 ng/ml, which are target concentrations predicted from preclinical data to inhibit receptor phosphorylation
in vitro and result in tumour stasis/growth reduction
in vivo. The primary active metabolite comprises 23% to 37% of the total exposure. No significant changes in the PK of sunitinib or the primary active metabolite are observed with repeated daily administration or with repeated cycles in the dosing schedules tested.
Absorption: After oral administration of sunitinib, Cmax are generally observed from 6 to 12 hours time to maximum concentration (tmax) post administration.
Food has no effect on the bioavailability of sunitinib.
Distribution: In vitro, binding of sunitinib and its primary active metabolite to human plasma protein was 95% and 90%, respectively, with no apparent concentration dependence. The apparent volume of distribution (Vd) for sunitinib was large, 2230 L, indicating distribution into the tissues.
Metabolic interactions: The calculated
in vitro Ki values for all cytochrome P450 (CYP) isoforms tested (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5, and CYP4A9/11) indicated that sunitinib and its primary active metabolite are unlikely to induce metabolism, to any clinically relevant extent, of other actives substances that may be metabolised by these enzymes.
Biotransformation: Sunitinib is metabolised primarily by CYP3A4, the CYP isoform which produces its primary active metabolite, desethyl sunitinib, which is then further metabolised by the same isoenzyme.
Co-administration of sunitinib with potent CYP3A4 inducers or inhibitors should be avoided because the plasma levels of sunitinib may be altered (see Precautions and Interactions).
Elimination: Excretion is primarily via faeces (61%), with renal elimination of unchanged active substance and metabolites accounting for 16% of the administered dose. Sunitinib and its primary active metabolite were the major compounds identified in plasma, urine, and faeces, representing 91.5%, 86.4%, and 73.8% of radioactivity in pooled samples, respectively. Minor metabolites were identified in urine and faeces, but generally were not found in plasma. Total oral clearance (CL/F) was 34-62 L/h. Following oral administration in healthy volunteers, the elimination half-lives of sunitinib and its primary active desethyl metabolite are approximately 40-60 hours and 80-110 hours, respectively.
Co-administration with medicinal products that are BCRP inhibitors: In vitro, sunitinib is a substrate of the efflux transporter BCRP. In study A6181038 the co-administration of gefitinib, a BCRP inhibitor, did not result in a clinically relevant effect on the Cmax and AUC for sunitinib or total drug (sunitinib + metabolite) (see Interactions). This study was a multicentre, open-label, Phase
1/
2 study examining the safety/tolerability, the maximum tolerated dose, and the antitumour activity of sunitinib in combination with gefitinib in subjects with MRCC. The PK of gefitinib (250 mg daily) and sunitinib (37.5 mg [Cohort 1, n=4] or 50 mg [Cohort 2, n=7] daily on a 4-weeks on followed by 2 weeks-off schedule) when co-administered was evaluated as a secondary study objective. Changes in sunitinib PK parameters were of no clinical significance and did not indicate any drug-drug interactions; however, considering the relatively low number of subjects (i.e. N=7+4) and the moderate-large interpatient variability in the pharmacokinetic parameters, caution needs to be taken when interpreting the PK drug-drug interaction findings from this study.
Special populations: Hepatic impairment: Sunitinib and its primary metabolite are mainly metabolised by the liver. Systemic exposures after a single dose of sunitinib were similar in subjects with mild or moderate (Child-Pugh Class A and B) hepatic impairment compared to subjects with normal hepatic function. ALVOSUNI was not studied in subjects with severe (Child-Pugh Class C) hepatic impairment.
Studies in cancer patients have excluded patients with ALT or AST >2.5 x ULN (upper limit of normal) or >5.0 x ULN if due to liver metastasis.
Renal impairment: Population PK analyses indicated that sunitinib apparent clearance (CL/F) was not affected by creatinine clearance (Clcr) within the range evaluated (42-347 ml/min). Systemic exposures after a single dose of sunitinib were similar in subjects with severe renal impairment (CLcr <30 ml/min) compared to subjects with normal renal function (CLcr >80 ml/min). Although sunitinib and its primary metabolite were not eliminated through haemodialysis in subjects with ESRD, the total systemic exposures were lower by 47% for sunitinib and 31% for its primary metabolite compared to subjects with normal renal function.
Weight, performance status: Population PK analyses of demographic data indicate that no starting dose adjustments are necessary for weight or Eastern Cooperative Oncology Group (ECOG) performance status.
Gender: Available data indicate that females could have about 30% lower apparent clearance (CL/F) of sunitinib than males: this difference, however, does not necessitate starting dose adjustments.
Paediatric population: Experience on the use of sunitinib in paediatric patients is limited (see Dosage & Administration). Population PK analyses of a pooled dataset from adult patients with GIST and solid tumours and paediatric patients with solid tumours were completed. Stepwise covariate modelling analyses were performed to evaluate the effect of age and body size (total body weight or body surface area) as well as other covariates on important PK parameters for sunitinib and its active metabolite. Among age and body size related covariates tested, age was a significant covariate on apparent clearance of sunitinib (the younger the age of the paediatric patient, the lower the apparent clearance). Similarly, body surface area was a significant covariate on the apparent clearance of the active metabolite (the lower the body surface area, the lower the apparent clearance).
Furthermore, based on an integrated population PK analysis of pooled data from the 3 paediatric studies (2 paediatric solid tumor studies and 1 paediatric GIST study; ages: 6 years to 11 years and 12 years to 17 years), baseline body surface area (BSA) was a significant covariate on apparent clearance of sunitinib and its active metabolite. Based on this analysis, a dose of approximately 20 mg/m
2 daily in paediatric patients, with BSA values between 1.10 and 1.87 m
2, is expected to provide plasma exposures to sunitinib and its active metabolite comparable (between 75 and 125% of the AUC) to those in adults with GIST administered sunitinib 50 mg daily on Schedule 4/2 (AUC 1233 ng·hr/mL). In paediatric studies, the starting dose of sunitinib was 15 mg/m
2 (based on the MTD identified in the Phase 1 dose escalation study, see PHARMACOLOGY: Pharmacodynamics under Actions), which in paediatric patients with GIST increased to 22.5 mg/m
2 and subsequently to 30 mg/m
2 (not to exceed the total dose of 50 mg/day) based on individual patient safety/tolerability. Furthermore, according to the published literatures in paediatric patients with GIST, the calculated starting dose ranged from 16.6 mg/m
2 to 36 mg/m
2, increased to doses as high as 40.4 mg/m
2 (not exceeding the total dose of 50 mg/day).
Toxicology: Preclinical safety data: In rat and monkey repeated-dose toxicity studies up to 9-months duration, the primary target organ effects were identified in the gastrointestinal tract (emesis and diarrhoea in monkeys); adrenal gland (cortical congestion and/or haemorrhage in rats and monkeys, with necrosis followed by fibrosis in rats); haemolymphopoietic system (bone marrow hypocellularity and lymphoid depletion of thymus, spleen, and lymph node); exocrine pancreas (acinar cell degranulation with single cell necrosis); salivary gland (acinar hypertrophy); bone joint (growth plate thickening); uterus (atrophy); and ovaries (decreased follicular development). All findings occurred at clinically relevant sunitinib plasma exposure levels. Additional effects observed in other studies included: QTc interval prolongation, LVEF reduction and testicular tubular atrophy, increased mesangial cells in kidney, haemorrhage in gastrointestinal tract and oral mucosa, and hypertrophy of anterior pituitary cells. Changes in the uterus (endometrial atrophy) and bone growth plate (physeal thickening or dysplasia of cartilage) are thought to be related to the pharmacological action of sunitinib. Most of these findings were reversible after 2 to 6 weeks without treatment.
Genotoxicity: The genotoxic potential of sunitinib was assessed
in vitro and
in vivo. Sunitinib was not mutagenic in bacteria using metabolic activation provided by rat liver. Sunitinib did not induce structural chromosome aberrations in human peripheral blood lymphocyte cells
in vitro. Polyploidy (numerical chromosome aberrations) was observed in human peripheral blood lymphocytes
in vitro, both in the presence and absence of metabolic activation. Sunitinib was not clastogenic in rat bone marrow
in vivo. The major active metabolite was not evaluated for genotoxic potential.
Carcinogenicity: In a 1-month, oral gavage dose-range finding study (0, 10, 25, 75, or 200 mg/kg/day) with continuous daily dosing in rasH2 transgenic mice, carcinoma and hyperplasia of Brunner's glands of the duodenum were observed at the highest dose (200 mg/kg/day) tested.
A 6-month, oral gavage carcinogenicity study (0, 8, 25, 75 [reduced to 50] mg/kg/day), with daily dosing was conducted in rasH2 transgenic mice. Gastroduodenal carcinomas, an increased incidence of background haemangiosarcomas, and/or gastric mucosal hyperplasia were observed at doses of ≥25 mg/kg/day following 1- or 6-months duration (≥7.3 times the AUC in patients administered the recommended daily dose [RDD]).
In a 2-year rat carcinogenicity study (0, 0.33, 1, or 3 mg/kg/day), administration of sunitinib in 28-day cycles followed by 7-day dose-free periods resulted in increases in the incidence of phaeochromocytomas and hyperplasia in the adrenal medulla of male rats given 3 mg/kg/day following >1 year of dosing (≥7.8 times the AUC in patients administered the RDD). Brunner's glands carcinoma occurred in the duodenum at ≥1 mg/kg/day in females and at 3 mg/kg/day in males, and mucous cell hyperplasia was evident in the glandular stomach at 3 mg/kg/day in males, which occurred at ≥0.9, 7.8, and 7.8 times the AUC in patients administered the RDD, respectively. The relevance to humans of the neoplastic findings observed in the mouse (rasH2 transgenic) and rat carcinogenicity studies with sunitinib treatment is unclear.
Reproductive and developmental toxicity: No effects on male or female fertility were observed in reproductive toxicity studies. However, in repeated-dose toxicity studies performed in rats and monkeys, effects on female fertility were observed in the form of follicular atresia, degeneration of corpora lutea, endometrial changes in the uterus, and decreased uterine and ovarian weights at clinically relevant systemic exposure levels. Effects on male fertility in rat were observed in the form of tubular atrophy in the testes, reduction of spermatozoa in epididymides, and colloid depletion in prostate and seminal vesicles at plasma exposure levels 25 times the systemic exposure in humans.
In rats, embryo-foetal mortality was evident as significant reductions in the number of live foetuses, increased numbers of resorptions, increased postimplantation loss, and total litter loss in 8 of 28 pregnant females at plasma exposure levels 5.5 times the systemic exposure in humans. In rabbits, reductions in gravid uterine weights and number of live foetuses were due to increases in the number of resorptions, increases in postimplantation loss and complete litter loss in 4 of 6 pregnant females at plasma exposure levels 3 times the systemic exposure in humans. Sunitinib treatment in rats during organogenesis resulted in developmental effects at ≥5 mg/kg/day consisting of increased incidence of foetal skeletal malformations, predominantly characterised as retarded ossification of thoracic/lumbar vertebrae and occurred at plasma exposure levels 5.5 times the systemic exposure in humans. In rabbits, developmental effects consisted of increased incidence of cleft lip at plasma exposure levels approximately equal to that observed in clinic, and cleft lip and cleft palate at plasma exposure levels 2.7 times the systemic exposure in humans.
Sunitinib (0.3, 1.0, 3.0 mg/kg/day) was evaluated in a pre-and postnatal development study in pregnant rats. Maternal body weight gains were reduced during gestation and lactation at ≥1 mg/kg/day but no maternal reproductive toxicity was observed up to 3 mg/kg/day (estimate exposure ≥2.3 times the AUC in patients administered the RDD). Reduced offspring body weights were observed during the preweaning and postweaning periods at 3 mg/kg/day. No development toxicity was observed at 1 mg/kg/day (approximate exposure ≥0.9 times the AUC in patients administered the RDD).


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image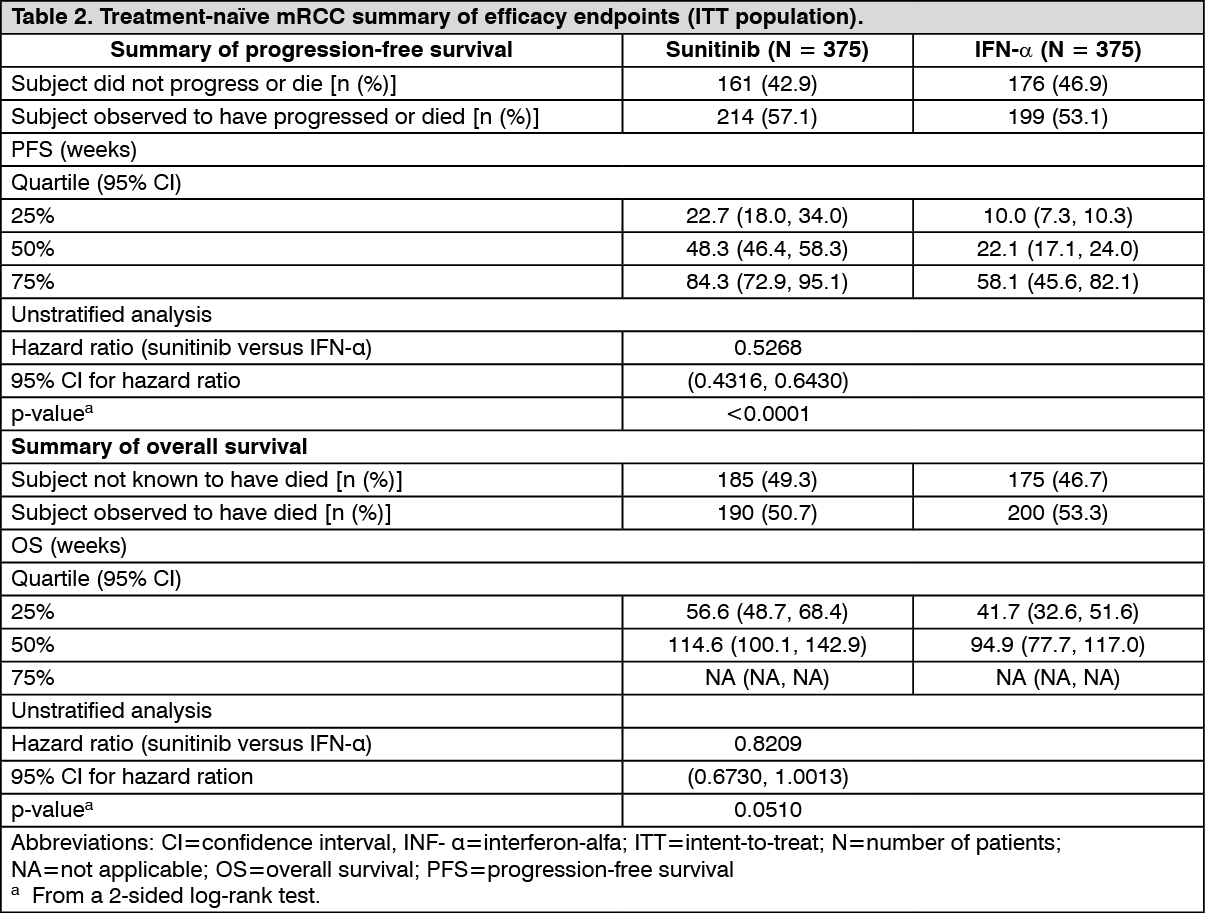 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image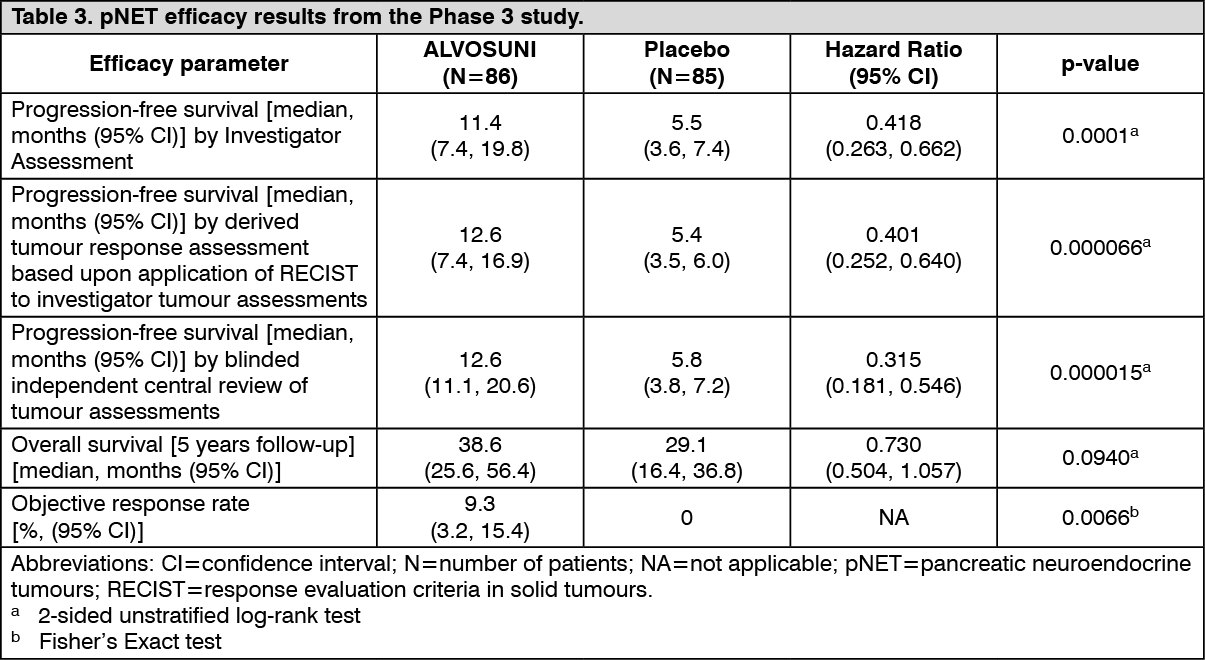 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image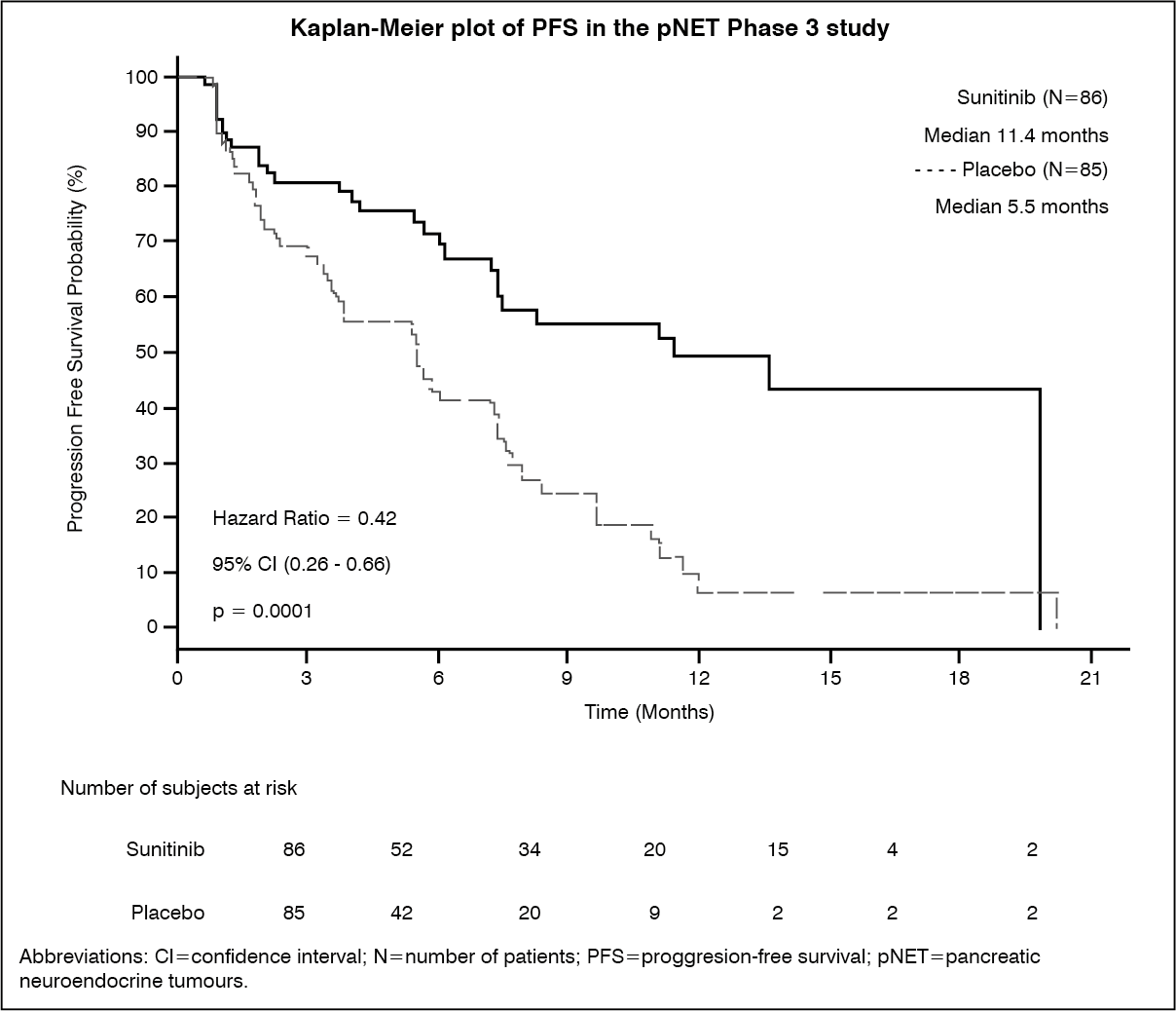 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image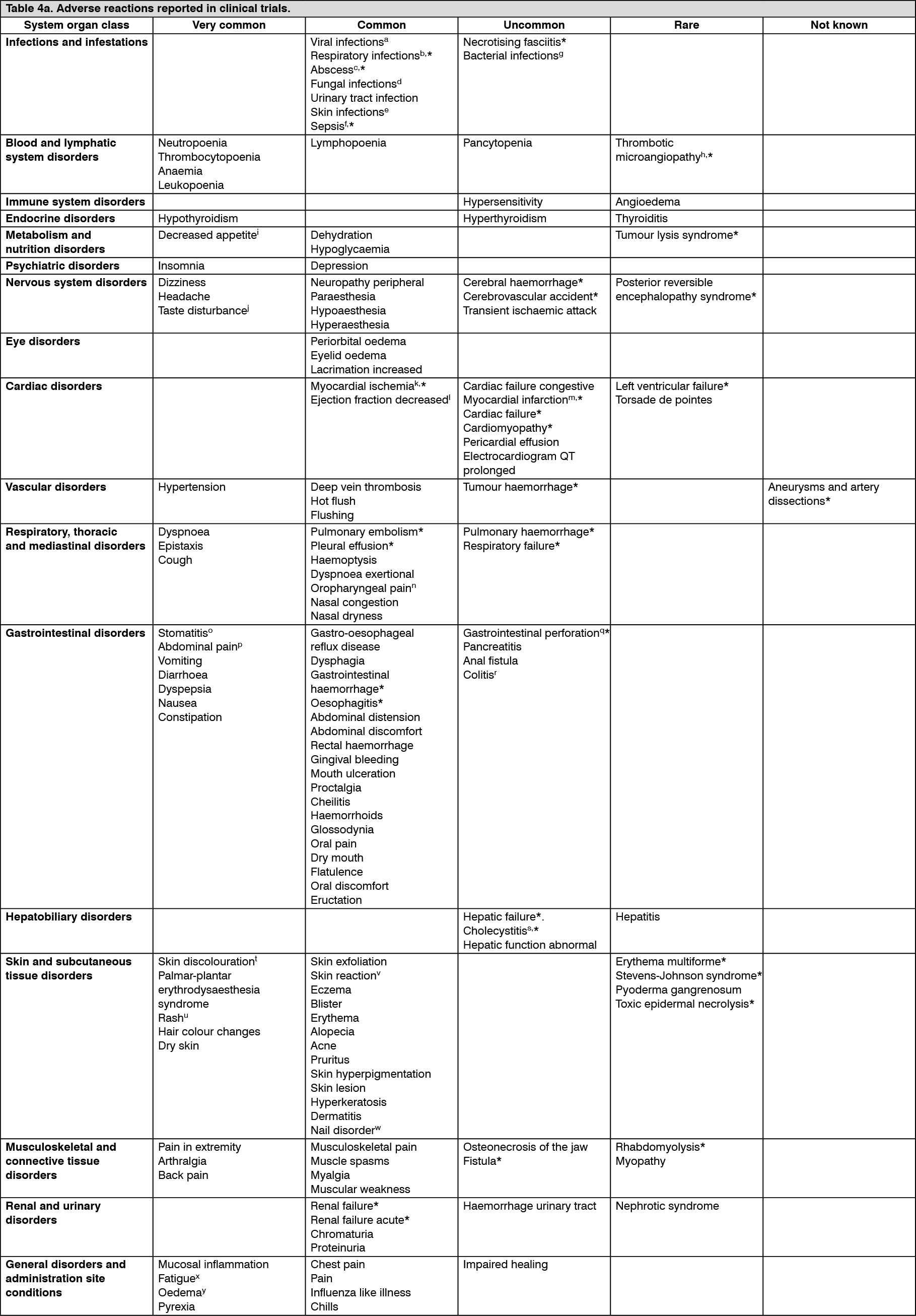 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image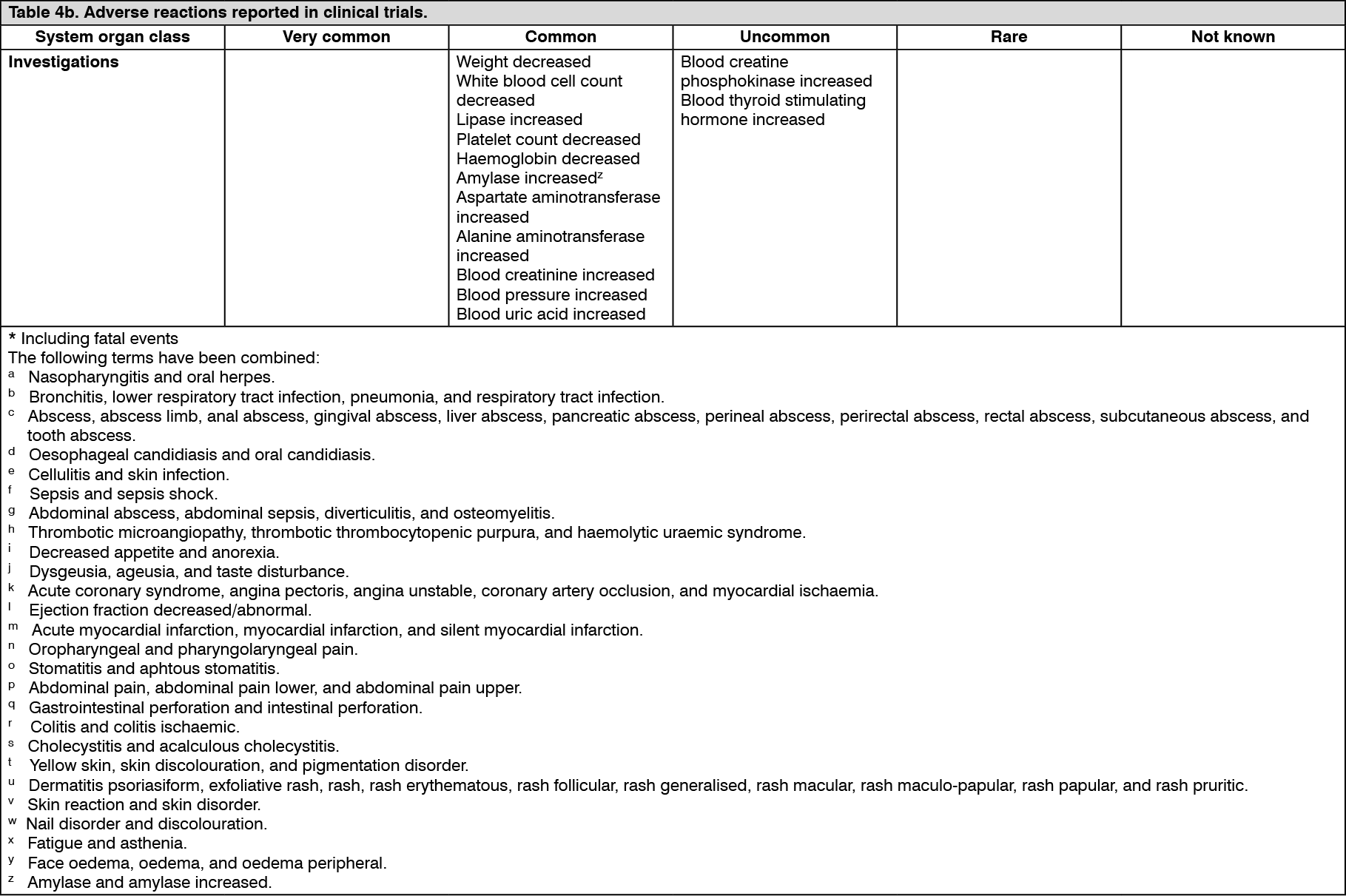 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
