MODE OF ADMINISTRATION: Capecitabine tablets should be swallowed whole with water within 30 minutes after a meal. Capecitabine tablets should not be crushed or cut. If patients cannot swallow capecitabine tablets whole and tablets must be crushed or cut, this should be done by a professional trained in the safe handling of cytotoxic drugs (see
Special instruction for use, handling and disposal under Cautions for Usage).
RECOMMENDED DOSE: Monotherapy: Colon, colorectal and breast cancer: The recommended monotherapy starting dose of capecitabine is 1250 mg/m
2 administered twice daily (morning and evening; equivalent to 2500 mg/m
2 total daily dose) for 2 weeks followed by a 7-day rest period.
Combination therapy: Breast cancer: In combination with docetaxel, the recommended starting dose of capecitabine is 1250 mg/m
2 twice daily for 2 weeks followed by a 7-day rest period, combined with docetaxel at 75 mg/m
2 as a 1-hour intravenous infusion every 3 weeks.
Premedication, according to the docetaxel labeling, should be started prior to docetaxel administration for patients receiving the capecitabine plus docetaxel combination.
Colon, colorectal, gastric and oesophagogastric cancer: In combination treatment (except with irinotecan), the recommended starting dose of capecitabine is 800 to 1000 mg/m
2 administered twice daily for 2 weeks followed by a 7-day rest period, or 625 mg/m
2 twice daily when administered continuously.
For combination with irinotecan (XELIRI), the recommended starting dose of capecitabine is 800 mg/m
2 administered twice daily for 2 weeks followed by a 7-day rest period in combination with irinotecan 200 mg/m
2 on day 1 of each three week cycle.
The inclusion of bevacizumab in a combination regimen has no effect on the starting dose of capecitabine. Adjuvant treatment in patients with stage III colon cancer is recommended for a total of 6 months.
Premedication to maintain adequate hydration and anti-emesis according to the cisplatin or oxaliplatin product information should be started prior to cisplatin administration for patients receiving the capecitabine plus cisplatin or oxaliplatin combination.
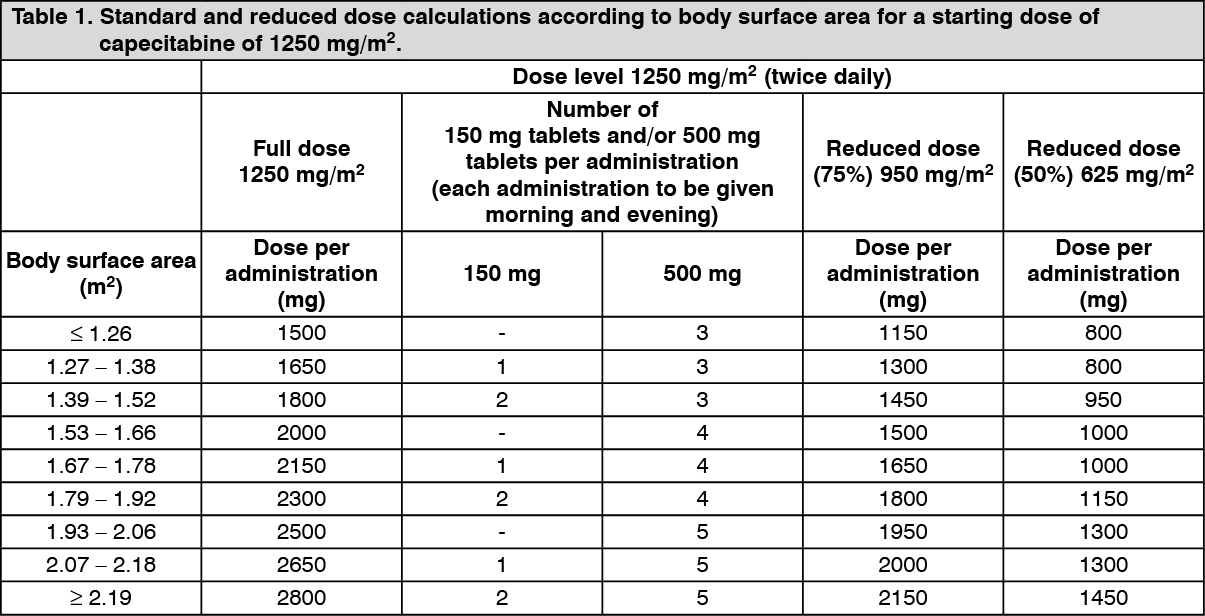
Dose calculation: Capecitabine dose is calculated according to body surface area. The following tables show examples of the standard and reduced dose calculations (see Dose adjustments during treatment as follows) for a starting dose of capecitabine of either 1250 mg/m
2 or 1000 mg/m
2. (See Tables 1 and 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Dose adjustments during treatment:
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Dose adjustments during treatment: General: Toxicity due to capecitabine administration may be managed by symptomatic treatment and/or modification of the capecitabine dose (treatment interruption or dose reduction). Once the dose has been reduced it should not be increased at a later time.
For those toxicities considered by the treating physician to be unlikely to become serious or life-threatening treatment can be continued at the same dose without reduction or interruption.
Dosage modifications are not recommended for Grade 1 events. Therapy with capecitabine should be interrupted if a Grade 2 or 3 adverse drug reaction (ADR) occurs. Once the ADR has resolved or decreased in intensity to Grade 1, capecitabine therapy may be restarted at full dose or as adjusted according to Table 3. If a Grade 4 ADR occurs, therapy should be discontinued or interrupted until the ADR has resolved or decreased to Grade 1, and therapy can then be restarted at 50% of the original dose. Patients taking capecitabine should be informed of the need to interrupt treatment immediately if moderate or severe toxicity occurs. Doses of capecitabine omitted for toxicity are not replaced.
Haematology: Patients with baseline neutrophil counts of <1.5x 10
9/L and/or thrombocyte counts of <100 x 10
9/L should not be treated with capecitabine. If unscheduled laboratory assessments during a treatment cycle show grade 3 or 4 haematologic toxicity, treatment with capecitabine should be interrupted.
The following table shows the recommended dose modifications following toxicity related to capecitabine. (See Table 3.)
Click on icon to see table/diagram/image
General combination therapy: Dose modifications for toxicity when capecitabine is used in combination with other therapies should be made according to Table 3 as previously mentioned for capecitabine and according to the appropriate prescribing information for the other agent(s).
At the beginning of a treatment cycle, if a treatment delay is indicated for either capecitabine or the other agent(s), then administration of all agents should be delayed until the requirements for restarting all drugs are met.
During a treatment cycle for those toxicities considered by the treating physician not to be related to capecitabine, capecitabine should be continued and the dose of the other agent adjusted according to the appropriate prescribing information.
If the other agent(s) have to be discontinued permanently, capecitabine treatment can be resumed when the requirements for restarting capecitabine are met.
This advice is applicable to all indications and to all special populations.
Special dosage instructions: Pediatric and adolescent use: The safety and efficacy of capecitabine in children and adolescents (<18 years) have not been established.
Geriatric use: For capecitabine monotherapy, no adjustment of the starting dose is needed. However, severe Grade 3 or 4 treatment-related ADRs were more frequent in patients over 80 years of age compared to younger patients.
When capecitabine was used in combination with other antineoplastic agents, geriatric patients (≥65 years) experienced more Grade 3 and Grade 4 ADRS and ADRs that led to discontinuation, than younger patients. Careful monitoring of elderly patients is advisable.
In combination with docetaxel: an increased incidence of Grade 3 or 4 treatment-related ADRs and treatment-related serious ADRS was observed in patients 60 years of age or more. For patients 60 years of age or more treated with the combination of capecitabine plus docetaxel, a starting dose reduction of capecitabine to 75% (950 mg/m
2 twice daily) is recommended. For dosage calculations, see Table 1.
Renal impairment: In patients with moderate renal impairment (creatinine clearance 30-50 mL/min [Cockroft and Gault]) at baseline, a dose reduction to 75% for a starting dose of 1250 mg/m
2 clearance 51-80 mL/min), no adjustment in starting dose is recommended. In patients with mild renal impairment (creatinine clearance 51-80 mL/min), no adjustment in starting dose is recommended.
Careful monitoring and prompt treatment interruption is recommended if the patient develops a Grade 2, 3 or 4 ADRS with subsequent dose adjustment as outlined in Table 3 as previously mentioned (see
PHARMACOLOGY: Pharmacokinetics under Actions). If the calculated creatinine clearance decreases during treatment to a value below 30 mL/min, capecitabine should be discontinued. The dose adjustment recommendations for patients with moderate renal impairment apply both to monotherapy and combination use. For dosage calculations, see Tables 1 and 2.
Hepatic impairment: In patients with mild to moderate hepatic impairment due to liver metastases, no starting dose adjustment is necessary. However, such patients should be carefully monitored (see
PHARMACOLOGY: Pharmacokinetics under Actions and
PRECAUTIONS). Patients with severe hepatic impairment have not been reported.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image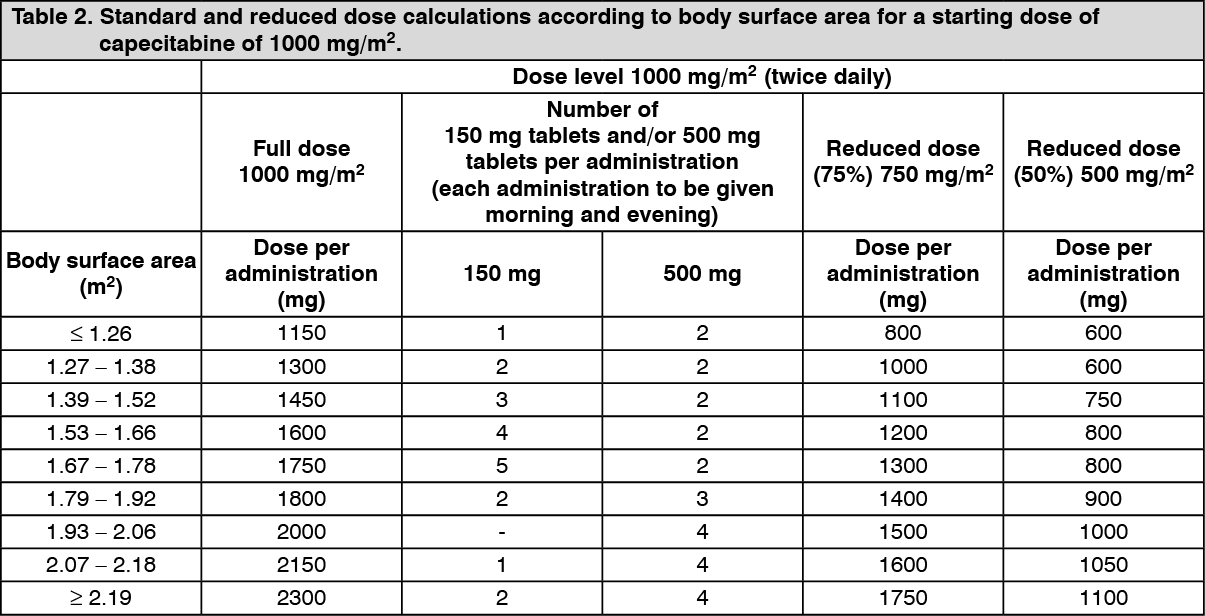 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image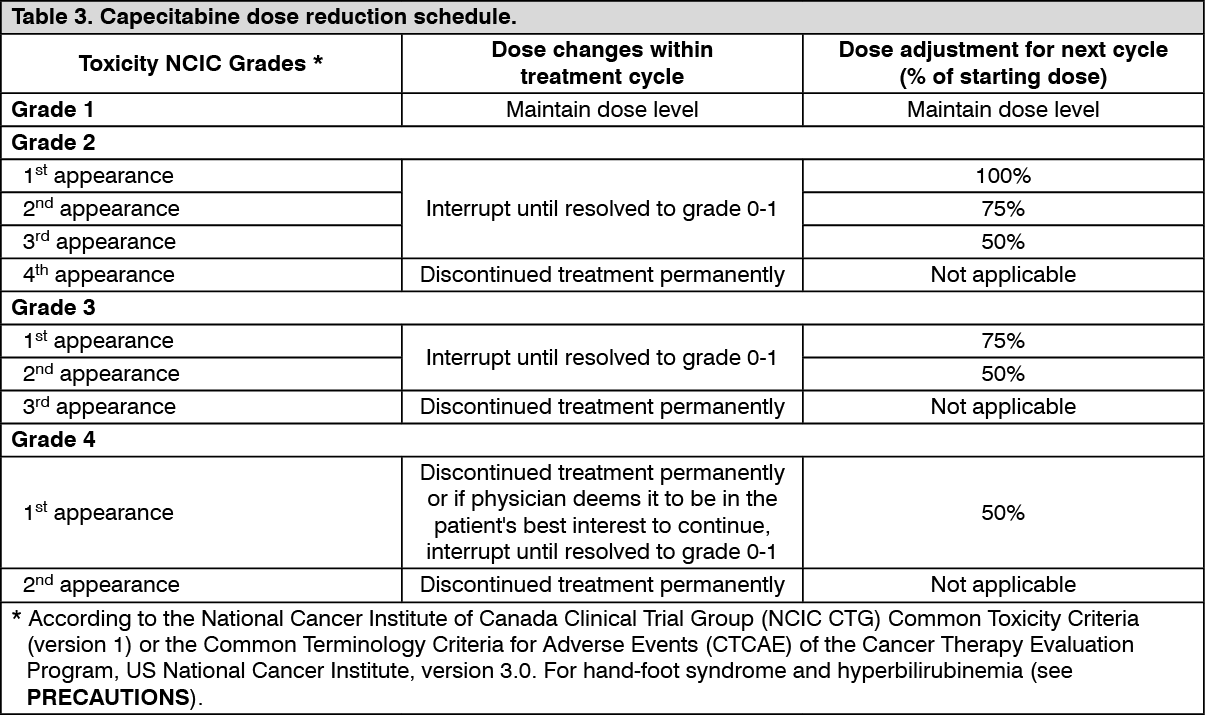 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out