


 Sign Out
Sign Out

Paracetamol is a non-opiate, non-salicylate analgesic.
Pharmacokinetics: Tramadol is administered as a racemate and both the (-) and (+) forms of both tramadol and M1 are detected in the circulation. The pharmacokinetics of plasma tramadol and paracetamol following oral administration of 1 tablet are shown in the Table as follows. Tramadol has a slower absorption and longer half-life when compared to paracetamol. (See Table.)
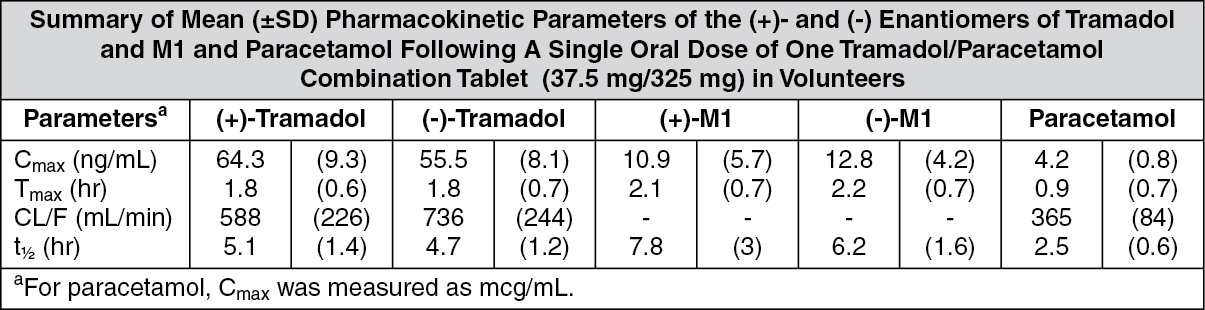 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAbsorption: Tramadol HCl has a mean absolute bioavailability of approximately 75% following administration of a single 100 mg oral dose of tramadol tablets. The mean peak plasma concentration of racemic tramadol and M1 after administration of 2 Doucetz occurs at approximately 2 and 3 hrs, respectively, post-dose.
Peak plasma concentrations of paracetamol occur within 1 hr and are not affected by co-administration with tramadol. Oral absorption of paracetamol following administration occurs primarily in the small intestine.
Distribution: The volume of distribution of tramadol was 2.6 and 2.9 L/kg in male and female subjects, respectively, following a 100 mg IV dose. The binding of tramadol to human plasma proteins is approximately 20% and binding also appears to be independent of concentration up to 10 mcg/mL. Saturation of plasma protein binding occurs only at concentrations outside the clinically relevant range.
Paracetamol appears to be widely distributed throughout most body tissues except fat. Its apparent volume of distribution is about 0.9 L/kg. A relative small portion (approximately 20%) of paracetamol is bound to plasma protein.
Metabolism: Following oral administration, tramadol is extensively metabolized by a number of pathways, including CYP2D6 and CYP3A4, as well as by conjugation of parent and metabolites.
Approximately 30% of the dose is excreted in the urine as unchanged drug, whereas 60% of the dose is excreted as metabolites. The major metabolic pathways appear to be N- and O- demethylation and glucuronidation or sulfation in the liver. Metabolite M1 (O-desmethyltramadol) is pharmacologically active in animal models. Formation of M1 is dependent on CYP2D6 and as such is subject to inhibition, which may affect the therapeutic response.
Approximately 7% of the population has reduced activity of the CYP2D6 isoenzyme of cytochrome P-450. These individuals are “poor metabolizers” of debrisoquine, dextromethorphan, tricyclic antidepressants, among other drugs. Based on a population PK analysis of phase 1 studies in healthy subjects, concentrations of tramadol were approximately 20% higher in “poor metabolizers” versus “extensive metabolizers”, while M1 concentrations were 40% lower. In vitro drug interaction studies in human liver microsomes indicates that inhibitors of CYP2D6 eg, fluoxetine and its metabolite norfluoxetine, amitriptyline and quinidine inhibit the metabolism of tramadol to various degrees. The full pharmacological impact of these alterations in terms of either efficacy or safety is unknown. Concomitant use of serotonin re-uptake inhibitors (SRIs) and monoamine oxidase inhibitors (MAOIs) may enhance the risk of adverse events, including seizure and serotonin syndrome.
Paracetamol is primarily metabolized in the liver by 1st-order kinetics and involves 3 principal separate pathways: Conjugation with glucuronide; conjugation with sulfate; and oxidation via the cytochrome P-450-dependent, mixed-function oxidase enzyme pathway to a reactive intermediate metabolite, which conjugates with glutathione and is then further metabolized to form cysteine and mercapturic acid conjugates. The principal cytochrome P-450 isoenzyme involved appears to be CYP2E1, with CYP1A2 and CYP3A4 as additional pathways.
In adults, the majority of paracetamol is conjugated with glucuronic acid and, to a lesser extent, with sulfate. These glucuronide-, sulfate- and glutathione-derived metabolites lack biologic activity. In premature infants, newborns and young infants, the sulfate conjugate predominates.
Elimination: Tramadol is eliminated primarily through metabolism by the liver and the metabolites are eliminated primarily by the kidneys. The plasma elimination half-lives of racemic tramadol and M1 are approximately 5-6 and 7 hrs, respectively, after administration of Duocetz. The apparent plasma elimination half-life of racemic tramadol increased to 7-9 hrs upon multiple dosing of Duocetz.
The half-life of paracetamol is about 2-3 hrs in adults. It is somewhat shorter in children and somewhat longer in neonates and in cirrhotic patients. Paracetamol is eliminated from the body primarily by formation of glucuronide and sulfate conjugates in a dose-dependent manner. Less than 9% of paracetamol is excreted unchanged in the urine.
Special Populations: Renal Impairment: The pharmacokinetics of Duocetz in patients with renal impairment has not been studied. Based on studies using tramadol alone, excretion of tramadol and metabolite M1 is reduced in patients with creatinine clearance of <30 mL/min, adjustment of dosing regimen in this patient population is recommended. The total amount of tramadol and M1 removed during a 4-hr dialysis period is <7% of the administered dose based on studies using tramadol alone.
Hepatic Impairment: The pharmacokinetics and tolerability of Duocetz in patients with impaired hepatic function has not been studied. Since tramadol and paracetamol are both extensively metabolized by the liver, the use of Duocetz in patients with hepatic impairment is not recommended.
Geriatric: A population pharmacokinetic analysis of data obtained from a clinical trial in patients with chronic pain treated with Duocetz which included 55 patients between 65 and 75 years and 19 patients >75 years, showed no significant changes in pharmacokinetics of tramadol and paracetamol in elderly patients with normal renal and hepatic function.
Gender: Tramadol clearance was 20% higher in female subjects compared to males on 4 phase I studies of Duocetz in 50 male and 34 female healthy subjects. The clinical significance of this difference is unknown.
Pediatric: Pharmacokinetics of Duocetz has not been studied in pediatric patients <16 years.
Toxicology: Carcinogenesis, Mutagenesis, Impairment of Fertility: There are no animal or laboratory studies on the Duocetz to evaluate carcinogenesis, mutagenesis or impairment of fertility.
Teratogenic Effects: No drug-related teratogenic effects were observed in the progeny of rats treated orally with tramadol and paracetamol. The Duocetz was shown to be embryotoxic and fetotoxic in rats at a maternally toxic dose, 50/434 mg/kg tramadol/paracetamol (300/2604 mg/m2 or 1.6 times the maximum daily human tramadol/paracetamol dosage of 185/1591 mg/m2), but was not teratogenic at this dose level. Embryo and fetal toxicity consisted of decreased fetal weights and increased supernumerary ribs.
Non-Teratogenic Effects: Tramadol alone was evaluated in peri- and postnatal studies in rats. Progeny of dams receiving oral (gavage) dose levels of 50 mg/kg (300 mg/m2 or 1.6 times the maximum daily human tramadol dosage) or greater had decreased weights, and pup survival was decreased early in lactation at 80 mg/kg (480 mg/m2 or 2.6 times the maximum daily human tramadol dosage).
There are no adequate and well-controlled studies in pregnant women. Duocetz should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. Neonatal seizures, neonatal withdrawal syndrome, fetal death and still birth have been reported with tramadol HCl during post-marketing.