


 Sign Out
Sign Out
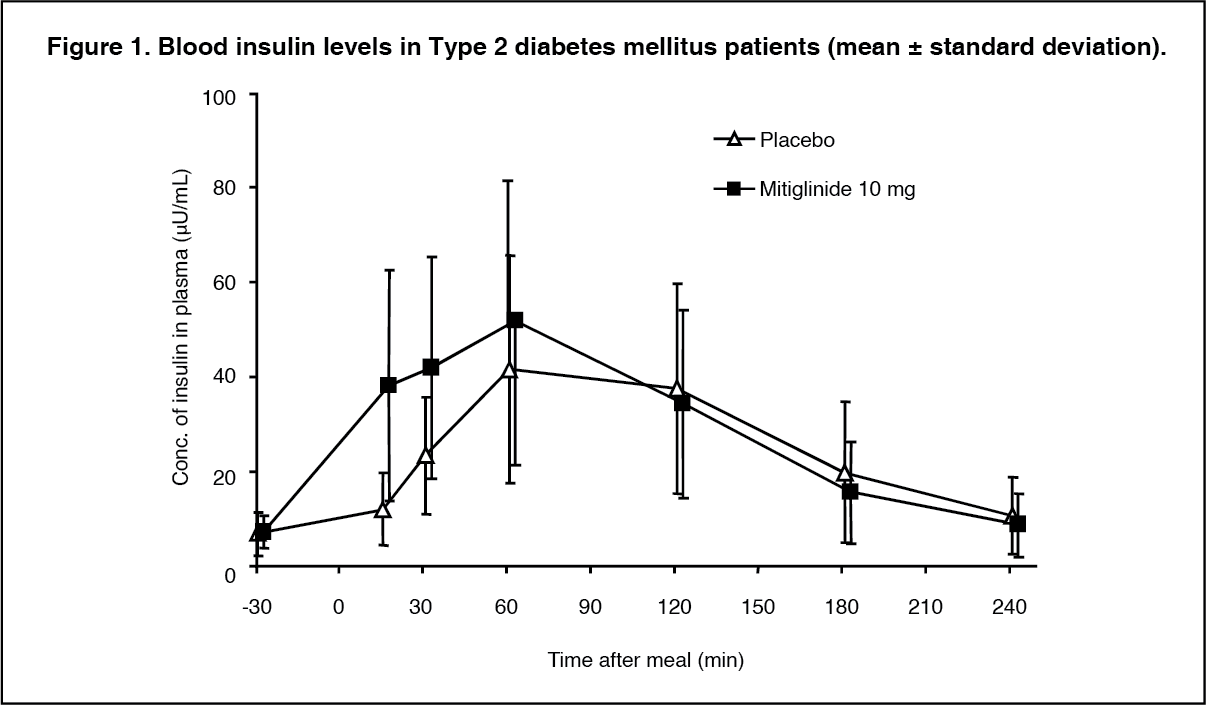 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image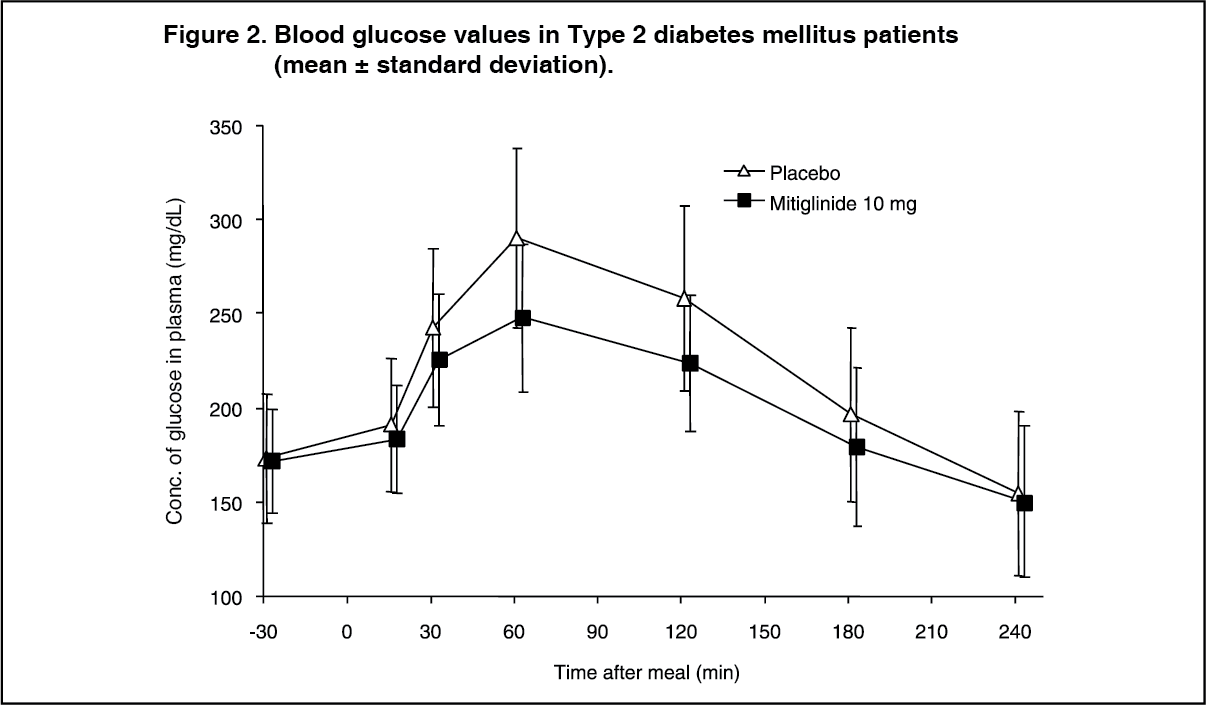 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageWhen mitiglinide calcium hydrate was administered orally to streptozotocin-induced diabetes mellitus model rats, because of its rapid-acting insulin secretion promoting activity, an increase in blood glucose after oral loading of liquid meal was suppressed, and post-loading plasma glucose area under the concentration-time curve value dropped (in vivo).
Mechanism of action: By bonding with pancreatic beta-cells' sulfonylurea receptors, mitiglinide calcium hydrate promotes the secretion of insulin by inhibiting the ATP-sensitive K+ channel (KATP channel) current (in vitro).
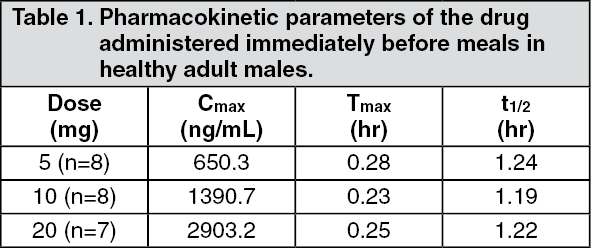
Pharmacokinetics: Plasma concentration: When a single dose of 5, 10 and 20 mg of mitiglinide was administered orally to healthy adult males immediately before meals, maximum plasma concentration (Cmax) was reached 0.23-0.28 hours after administration, and the half-life (t½) was approximately 1.2 hours. (See Table 1 and Figure 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image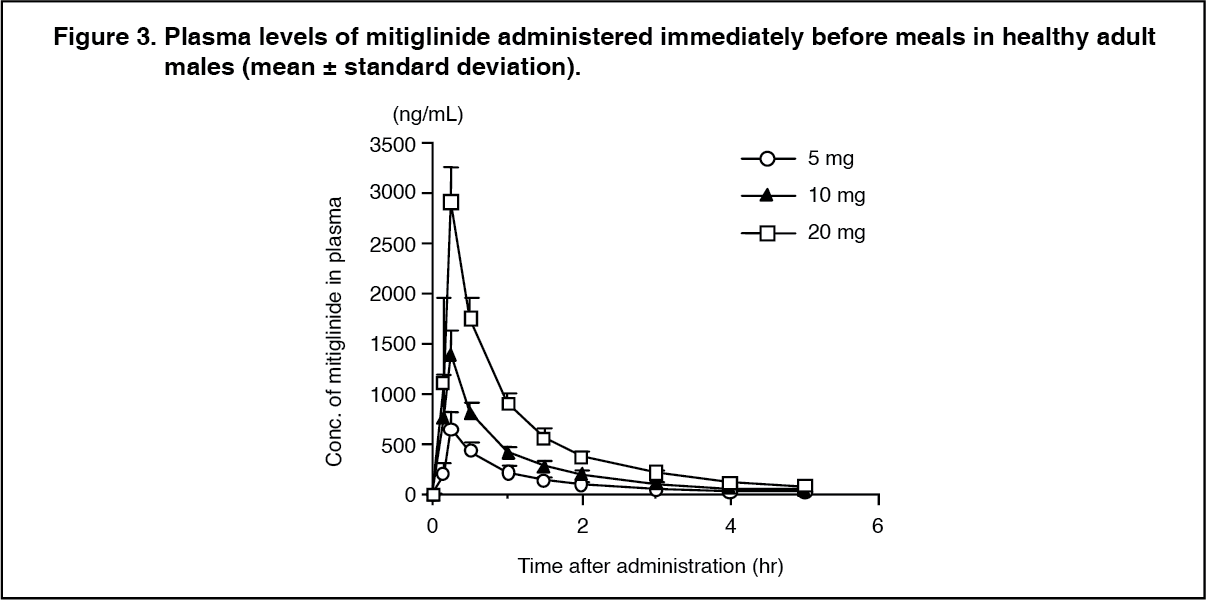 Click on icon to see table/diagram/image
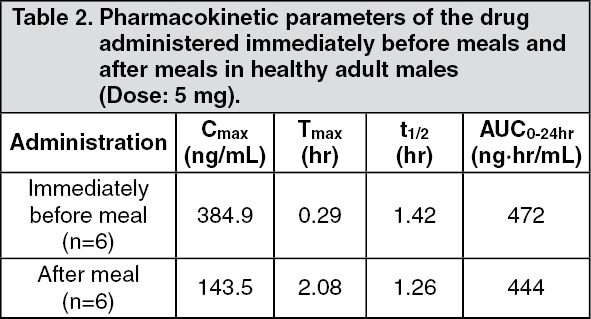
Click on icon to see table/diagram/imageOn the other hand, when 5 mg of mitiglinide was administered orally after meals to healthy adult males, compared with immediately before meals, a reduction in maximum plasma concentration (Cmax) and a delay in the time to reach maximum plasma concentration (Tmax) were observed. (See Table 2.)
 Click on icon to see table/diagram/image
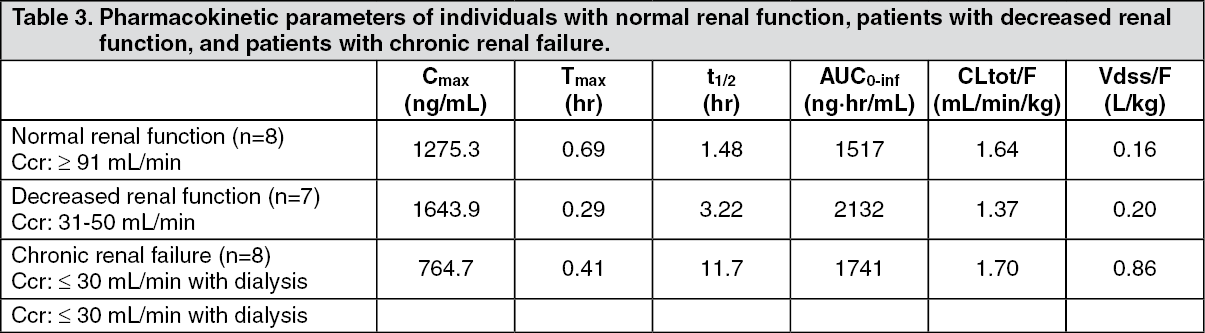
Click on icon to see table/diagram/imageWhen a single dose of 10 mg of mitiglinide was administered orally to adults with normal renal function, patients with decreased renal function, and patients with chronic renal failure (mean creatinine clearance values on the day before administration of this drug were 113.75, 37.01 and 3.431 mL/min, respectively) immediately before meals, along with a reduction in creatinine clearance, the half-life (t½) was prolonged. However, no significant correlation was seen between creatinine clearance and other major parameters (Cmax, AUC0-inf and CL tot/F). (See Table 3 and Figure 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image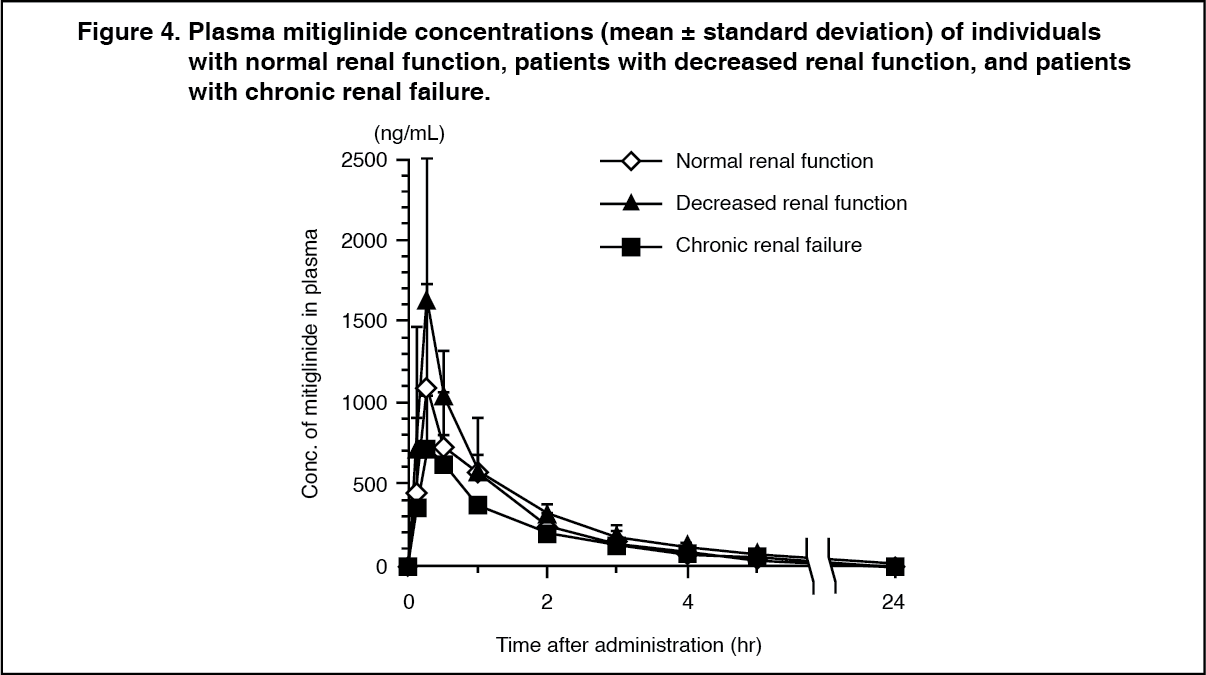 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageWhen a single dose of mitiglinide at 10 mg was administered concomitantly with voglibose (an α-glucosidase inhibitor) immediately before meal in patients with Type 2 diabetes mellitus whose blood glucose control by voglibose alone was inadequate, maximum plasma concentration (Cmax) of 1395.8 ng/mL was reached 0.28 hours after administration, and the half-life (t½) was 1.29 hours. Thus, no significant changes were observed in pharmacokinetic profiles compared with mitiglinide administration alone.
Metabolism and excretion: When a single dose of 5, 10 and 20 mg of mitiglinide was administered orally to healthy adult males immediately before meals, approximately 54-74% of the dose was excreted in the urine within 24 hours, a majority in the form of glucuronide conjugate metabolites. Mitiglinide accounted for less than 1%.
When a single dose of the 11 mg solution of 14C-labeled mitiglinide calcium hydrate was administered orally to non-Japanese healthy adult males immediately before meals, radioactivity in the plasma 0.5 hours and 4 hours after administration was primarily mitiglinide-derived, and the amount of mitiglinide's glucuronide conjugate that existed was approximately 1/3 to 1/6 the amount of mitiglinide. There were even fewer hydroxyl metabolites. Approximately 93% of the radioactivity was excreted in the urine, and approximately 6% in the feces.
In vitro studies have confirmed that mitiglinide calcium hydrate is metabolized in the liver and kidneys in humans, while glucuronide conjugates are mainly produced by the drug metabolic enzymes UGT1A9 and 1A3, and hydroxymetabolites, mainly by CYP2C9.
Clinical Studies: Monotherapy: Placebo-controlled double-blind phase II study: In 190 patients with Type 2 diabetes mellitus who are unable to obtain sufficient blood glucose control with diet therapy alone (44.4% had no history of drug therapy for diabetes mellitus; mean HbA1c value at the start of administration: 8.03%), a dose of 5, 10 and 20 mg of this drug was administered TID orally immediately before meals for 12 weeks. The patients in the 10-mg group showed a significant decrease in HbA1c eight weeks after administration. At the time of the final evaluation, the HbA1c variation was, compared with +0.49% in the placebo group, -0.22%, -0.35%, and -0.38% in the 5-, 10- and 20-mg group, respectively, showing significant reductions over the placebo in all dose groups (p<0.025, Shirley-Williams test). Frequencies of hypoglycemic symptoms, moreover, were 6.7%, 2.2%, and 6.3%, respectively, in the drug groups compared with 6.5% in the placebo group.
Placebo-controlled double-blind phase III study: In 314 patients with Type 2 diabetes mellitus who are unable to obtain sufficient blood glucose control with diet therapy alone (79.4% had no history of drug therapy for diabetes mellitus; mean HbA1c value at the start of administration: 7.47%), a dose of 10 mg of this drug was administered TID orally immediately before meals for 12 weeks. At the time of the final evaluation, the HbA1c variation was compared with +0.21% in the placebo group, -0.44% in the 10-mg group, showing a significant difference (p<0.001, t-test). Frequency of hypoglycemic symptoms, moreover, was 2.0% in the 10-mg group compared with 2.9% in the placebo group.
Long-term study: In the long-term administration study, with a dose of 10 mg, 218 out of 351 patients showed their HbA1c decreased, and stable blood glucose control was successfully maintained thereafter. Thirty-seven patients showed improvements in HbA1c with increased dose to 20 mg sixteen weeks after the start of administration because of failing to obtain effects with a dose of 10 mg.
Concomitant treatment with α-glucosidase inhibitors: Double-blind phase II/III study: In 385 patients with Type 2 diabetes mellitus whose blood glucose control was inadequate (mean HbA1c at the start of concomitant treatment: 7.10%) by the treatment regimen with diet and voglibose monotherapy (a single dose: 0.2 mg), voglibose at a dose of 0.2 mg and mitiglinide at a dose of 5 or 10 mg were orally administered 3 times daily immediately before meals for 12 weeks. The changes in HbA1c at the time of the final evaluation showed a significant decrease: -0.64% in the 10 mg mitiglinide treatment group and -0.44% in the 5 mg mitiglinide treatment group whereas the change was -0.02% in the voglibose monotherapy group (p<0.001, ANOVA for all groups).
Frequencies of hypoglycemic symptoms were 6.9% in the 10 mg mitiglinide treatment group and 3.3% in the 5 mg mitiglinide treatment group whereas the frequency was 1.1% in the voglibose monotherapy group.
Long-term concomitant treatment study: In 161 patients with Type 2 diabetes mellitus receiving long-term administration of mitiglinide concomitantly with voglibose, stable improvement in HbA1c was observed.