Pharmacotherapeutic Group: Antineoplastic agent.
ATC code: LO1 XC03.
Pharmacology: Pharmacodynamics: Mechanism of Action: Trastuzumab is a recombinant humanised monoclonal antibody that selectively targets the extracellular domain of the human epidermal growth factor receptor 2 protein (HER2). The antibody is an IgG, isotype that contains human framework regions with the complementarity-determining regions of a murine anti-p185 HER2 antibody that binds to human HER2.
The HER2 proto-oncogene or c-erbB2 encodes for a single transmembrane spanning, receptor-like protein of 185 kDa, which is structurally related to the epidermal growth factor receptor. Overexpression of HER2 is observed in 15%-20% of primary breast. The overall rate of HER2 positivity in advanced gastric cancers as observed during screening for study BO18255 is 15% for IHC3+ and IHC2+/FISH+ or 22.1% when applying the broader definition of IHC3+ or FISH+. A consequence of HER2 gene amplification is an increase in HER2 protein expression on the surface of these tumour cells, which results in a constitutively activated HER2 protein.
Studies indicate that breast cancer patients whose tumours have amplification or overexpression of HER2 have a shortened disease-free survival compared to patients whose tumours do not have amplification or overexpression of HER2.
Trastuzumab has been shown, both in in-vitro assays and in animals, to inhibit the proliferation of human tumour cells that overexpress HER2. In vitro, trastuzumab-mediated antibody-dependent cell-mediated cytotoxicity (ADCC) has been shown to be preferentially exerted on HER2 overexpressing cancer cells compared with cancer cells that do not overexpress HER2.
Clinical/ Efficacy Studies: Metastatic Breast Cancer: Trastuzumab monotherapy has been used in clinical trials for patients with metastatic breast cancer who have tumours that overexpress HER2 and who have failed one or more chemotherapy regimens for their metastatic disease.
Trastuzumab has also been used in clinical trials in combination with paclitaxel or an anthracycline (doxorubicin or epirubicin) + cyclophosphamide as first line therapy for patients with metastatic breast cancer who have tumours that overexpress HER2.
Patients who had previously received anthracycline-based adjuvant chemotherapy were treated with paclitaxel (175 mg/m
2 infused over 3 hours) with or without trastuzumab. Patients could be treated with trastuzumab until progression of disease.
Trastuzumab monotherapy, when used as second- or third-line treatment of women with metastatic breast cancer which overexpresses HER2, results in an overall tumour response rate of 15% and a median survival of 13 months. The use of trastuzumab in combination with paclitaxel as first-line treatment of women with metastatic breast cancer that overexpresses HER2 significantly prolongs the median time to disease progression, compared with patients treated with paclitaxel alone. The increase in median time to disease progression for patients treated with trastuzumab and paclitaxel is 3.9 months (6.9 months versus 3.0 months). Tumour response and one year survival rate are also increased for trastuzumab in combination with paclitaxel versus paclitaxel alone.
Trastuzumab has also been studied in a randomised, controlled trial, in combination with docetaxel, as first-line treatment of women with metastatic breast cancer. The combination of trastuzumab and docetaxel significantly increased response rate (61% versus 34%) and prolonged the median time to disease progression (by 5.6 months), compared with patients treated with docetaxel alone. Median survival was also significantly increased in patients receiving the combination, compared with those receiving docetaxel alone (31.2 months versus 22.7 months).
Combination treatment with trastuzumab and anastrozole: Trastuzumab has been studied in combination with anastrozole for first line treatment of metastatic breast cancer in HER2 overexpressing, hormone-receptor [i.e. oestrogen-receptor (ER) and/or progesterone-receptor (PR)] positive patients. Progression-free survival was doubled in the trastuzumab + anastrozole arm compared to anastrozole (4.8 months versus 2.4 months). For the other parameters the improvements seen for the combination were: for overall response (16.5% versus 6.7%); clinical benefit rate (42.7% versus 27.9%); time to progression (4.8 months versus 2.4 months). For time to response and duration of response no difference could be recorded between the arms. The median overall survival was extended by 4.6 months for patients in the combination arm. The difference was not statistically significant; however, more than half of the patients in the anastrozole alone arm crossed over to a trastuzumab containing regimen after progression of disease. Fifty two percent of the patients taking trastuzumab + anastrozole survived for at least 2 years compared to 45% taking anastrozole alone.
Early Breast Cancer: In the adjuvant setting, trastuzumab was investigated in 4 large multicentre, randomised, phase 3 trials: The Study BO16348 was designed to compare one and two years of three-weekly trastuzumab treatment versus observation in patients with HER2 positive early breast cancer following surgery, established chemotherapy and radiotherapy (if applicable). In addition, a comparison of two years trastuzumab treatment versus one year trastuzumab treatment was performed. Patients assigned to receive trastuzumab were given an initial loading dose of 8 mg/kg, followed by 6 mg/kg every three weeks for either one or two years.
Studies NSAPB B-31 and NCCTG N9831 that comprise the joint analysis were designed to investigate the clinical utility of combining trastuzumab treatment with paclitaxel following AC chemotherapy; additionally the NCCTG N9831 study investigated adding trastuzumab sequentially to AC-paclitaxel chemotherapy in patients with HER2-positive early breast cancer following surgery. Study BCIRG 006 was designed to investigate combining trastuzumab treatment with docetaxel either following AC chemotherapy or in combination with docetaxel and carboplatin in patients with HER2-positive early breast cancer following surgery.
Study BCIRG 006 was designed to investigate combining trastuzumab treatment with docetaxel either following AC chemotherapy or in combination with docetaxel and carboplatin in patients with HER2-positive early breast cancer following surgery.
Early breast cancer in the BO16348 study was limited to operable, primary, invasive adenocarcinoma of the breast, with axillary nodes-positive or axillary nodes-negative tumours of at least 1 cm in diameter.
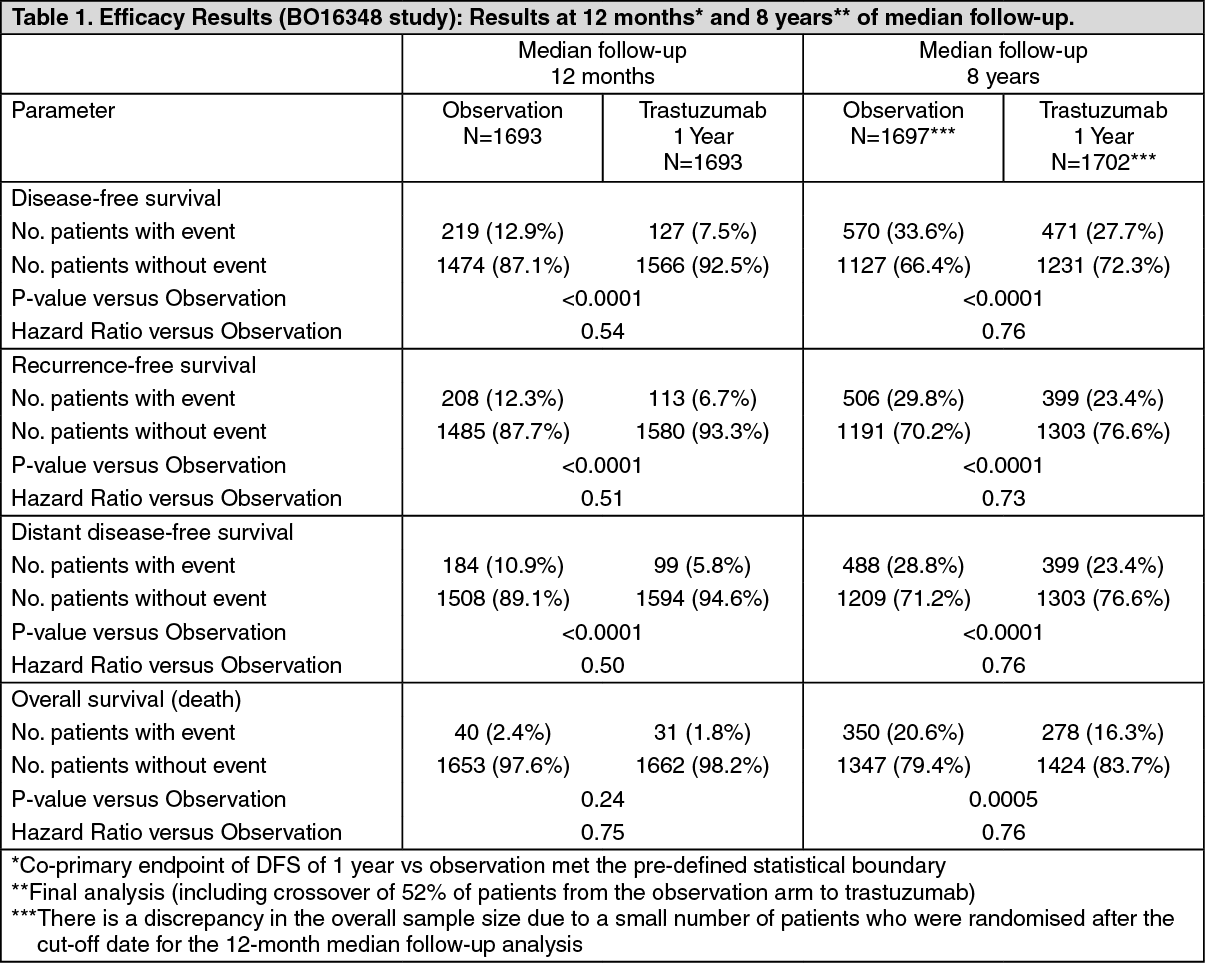
The efficacy results from the BO16348 study are summarized in the following table: See Table 1.
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The efficacy results from the interim efficacy analysis crossed the protocol pre-specified statistical boundary for the comparison of 1-year of trastuzumab vs. observation. After a median follow-up of 12 months, the hazard ratio (HR) for disease free survival (DFS) was 0.54 (95% CI 0.44, 0.67) which translates into an absolute benefit, in terms of a 2-year disease-free survival rate, of 7.6 percentage points (85.8% versus 78.2%) in favour of the trastuzumab arm.
A final analysis was performed after a median follow-up of 8 years, which showed that 1-year trastuzumab treatment is associated with a 24% risk reduction compared to observation only (HR=0.76, 95% CI 0.67, 0.86). This translates into an absolute benefit in terms of an 8-year disease free survival rate of 6.4 percentage points in favour of 1 year trastuzumab treatment.
In this final analysis, extending trastuzumab treatment for a duration of two years did not show additional benefit over treatment for 1 year [DFS HR in the intent to treat (ITT) population of 2 years versus 1 year=0.99 (95% CI: 0.87, 1.13), p-value = 0.90 and OS HR=0.98 (0.83, 1.15); p-value= 0.78].
The rate of asymptomatic cardiac dysfunction was increased in the 2-year treatment arm (8.1% versus 4.6% in the 1-year treatment arm). More patients experienced at least one Grade 3 or 4 adverse event in the 2-year treatment arm (20.4%) compared with the 1-year treatment arm (16.3%). In the joint analysis of the NSAPB B-31 and NCCTG N9831 studies, early breast cancer was limited to women with operable breast cancer at high risk, defined as HER2-positive and axillary lymph node-positive or HER2-positive and lymph node-negative with high risk features (tumour size > 1 cm and ER-negative or tumour size > 2 cm, regardless of hormonal status). Trastuzumab was administered in combination with paclitaxel, following AC chemotherapy. Paclitaxel was administered as follows: intravenous paclitaxel - 80 mg/m
2 as a continuous IV infusion, given every week for 12 weeks, or intravenous paclitaxel - 175 mg/m
2 as a continuous IV infusion, given every 3 weeks for 4 cycles (day 1 of each cycle). (See Table 2.)
Click on icon to see table/diagram/image
For the primary endpoint, DFS, the addition of trastuzumab to paclitaxel chemotherapy resulted in a 52% decrease in the risk of disease recurrence. The hazard ratio translates into an absolute benefit, in terms of a 3-year disease-free survival rate, of 11.8 percentage points (87.2% versus 75.4%) in favour of the AC→PH (trastuzumab) arm.
The pre-planned final analysis of OS from the joint analysis of studies NSABP B-31 and NCCTG N9831 was performed when 707 deaths had occurred (median follow-up 8.3 years in the AC→PH group). Treatment with AC→PH resulted in a statistically significant improvement in OS compared with AC->P (stratified HR=0.64; 95% CI [0.55, 0.74); log-rank p-value < 0.0001). At 8 years, the survival rate was estimated to be 86.9% in the AC→PH arm and 79.4% in the AC→P arm, an absolute benefit of 7.4% (95% CI 4.9%, 10.0%).
The final OS results from the joint analysis of studies NSABP B-31 and NCCTG N9831 are summarized in the following table: See Table 3.
Click on icon to see table/diagram/image
In the BCIRG 006 study, HER2-positive, early breast cancer was limited to either lymph node-positive or high risk node-negative patients, defined as negative (pN0) lymph node involvement, and at least 1 of the following factors: tumour size greater than 2 cm, oestrogen receptor- and progesterone receptor-negative, histologic and/or nuclear grade 2 - 3, or age < 35 years.
Trastuzumab was administered either in combination with docetaxel, following AC chemotherapy (AC-DH) or in combination with docetaxel and carboplatin (DCarbH). Docetaxel was administered as follows: intravenously (100 mg/m
2 as an IV infusion over 1 hour) given every 3 weeks for 4 cycles (day 2 of first docetaxel cycle, then day 1 of each subsequent cycle), or intravenously (75 mg/m
2 as an IV infusion over 1 hour) given every 3 weeks for 6 cycles (day 2 of cycle 1, then day 1 of each cycle).
Docetaxel therapy was followed by carboplatin (at target AUC = 6 mg/ml/min) administered by IV infusion over 30-60 minutes repeated every 3 weeks for a total of 6 cycles.
The efficacy results from the BCIRG 006 study are summarized in the following tables: See Tables 4 and 5.
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
In the BCIRG 006 study for the primary endpoint, DFS, the hazard ratio translates into an absolute benefit, in terms of a 3-year disease-free survival rate, of 5.8 percentage points (86.7% versus 80.9%) in favour of the AC→DH (trastuzumab) arm and 4.6 percentage points (85.5% versus 80.9%) in favour of the DCarbH (trastuzumab) arm compared to AC→D.
For the secondary endpoint overall survival, treatment with AC→DH reduced the risk of death by 42% when compared to AC→D (hazard ratio 0.58 [95% CI: 0.40, 0.83], p = 0.0024, log-rank test), and the risk of death was reduced by 34% for patients treated with DCarbl-1 compared to patients treated with AC→D (hazard ratio 0.66 [95% CI: 0.47, 0.93], p = 0.0182). In the BCIRG 006 study at the second interim analysis, 185 randomised patients had died: 80 patients (7.5%) in the AC→D arm, 49 patients (4.6%) in the AC→DH arm, and 56 patients (5.2%) in the DCarbH arm. The median duration of follow-up was 2.9 years in the AC→D arm and 3.0 years in both the AC→DH and DCarbH arms.
In the neoadjuvant-adjuvant setting, trastuzumab was evaluated in two phase 3 trials.
Study MO16432 investigated a total of 10 cycles of neoadjuvant chemotherapy [an anthracycline and a taxane (AP+H followed by P+H, followed by CMF+H] concurrently with neoadjuvant-adjuvant trastuzumab, or neoadjuvant chemotherapy alone, followed by adjuvant trastuzumab for up to a total treatment duration of 1 year) in newly diagnosed locally advanced (Stage III) or inflammatory HER2 positive breast cancer patients.
The efficacy results from MO16432 are summarized in the table as follows. The median duration of follow-up in the trastuzumab arm was 3.8 years. (See Table 6.)
Click on icon to see table/diagram/image
For the primary endpoint, EFS, the addition of trastuzumab to the neoadjuvant chemotherapy followed by adjuvant trastuzumab for a total duration of 52 weeks resulted in a 35% reduction in the risk of disease recurrence/progression. The hazard ratio translates into an absolute benefit, in terms of 3-year event-free survival rate estimates of 13 percentage points (65% versus 52%) in favour of the trastuzumab arm.
Advanced Gastric Cancer: The efficacy results from the BO18255 study are summarized in table 7. Patients with previously untreated for HER2-positive inoperable locally advanced or recurrent and/or metastatic adenocarcinoma of the stomach or gastro-oesophageal junction not amenable to curative therapy were recruited. The primary endpoint was overall survival which was defined as the time from the date of randomization to the date of death from any cause. At the time of the analysis a total of 349 randomised patients had died: 182 patients (62.8%) in the control arm and 167 patients (56.8%) in the treatment arm. The majority of the deaths were due to events related to the underlying cancer.
The overall survival was significantly improved in the trastuzumab + capecitabine/5-FU and cisplatin arm compared to the capecitabine/5-FU and cisplatin arm (p = 0.0046, log-rank test). The median survival time was 11.1 months with capecitabine/5-FU and cisplatin and 13.8 months with trastuzumab + capecitabine/5-FU and cisplatin. The risk of death was decreased by 26% (hazard ratio [HR] 0.74 95% CI [0.60-0.91]) for patients in the trastuzumab arm compared to the capecitabine/5-FU arm. Post-hoc subgroup analyses indicate that targeting tumours with higher levels of HER2 protein (IHC 2+/FISH+ and IHC 3+/regardless of the FISH status) results in a greater treatment effect. The median overall survival for the high HER2-expressing group was 11.8 months versus 16 months, HR 0.65 (95% CI 0.51-0.83) and the median progression-free survival was 5.5 months versus 7.6 months, HR 0.64 (95% CI 0.51-0.79) for capecitabine/5-FU and cisplatin and trastuzumab + capecitabine/5-FU and cisplatin, respectively.
In a method comparison study a high degree of concordance (> 95%) was observed for SISH and FISH techniques for the detection of HER2 gene amplification in gastric cancer patients. (See Table 7.)
Click on icon to see table/diagram/image
Pharmacokinetics: The pharmacokinetics of trastuzumab was evaluated in a population pharmacokinetic model analysis using pooled data from 1,582 subjects from 18 Phase I, II and III trials receiving intravenous trastuzumab. A two-compartment model with parallel linear and non-linear elimination pathways from the central compartment was used to describe trastuzumab concentration-time profile. Due to the non-linear elimination pathway, total clearance increases with decreasing concentrations. Linear elimination clearance was 0.127 l/day for breast cancer (MBC/EBC) and 0.176 l/day for AGC. The nonlinear elimination parameters were 8.81 mg/day for the maximum elimination rate (V
max) and 8.92 mg/l for the Michaelis-Menten constant (K
m). The central compartment volume was 2.62 l for patients with breast cancer and 3.63 l for patients with AGC.
The population predicted PK exposures (with 5th - 95th Percentiles) and PK parameters at clinically relevant concentrations (C
max and C
min) for breast cancer and AGC patients following the approved Q1W and Q3W regimens are shown in Table 8 (Cycle 1) and Table 9 (steady-state) as follows. (See Tables 8 and 9.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Pharmacokinetics in Special Populations: Detailed pharmacokinetic studies in the elderly and those with renal or hepatic impairment have not been carried out.
Renal Impairment: Detailed pharmacokinetic studies in patients with renal impairment have not been carried out. In a population pharmacokinetic analysis, renal impairment was shown not to affect trastuzumab disposition.
Elderly: Age has been shown to have no effect on the disposition of trastuzumab (see Dosage & Administration).
Toxicology: Preclinical Safety: Trastuzumab was well tolerated in mice (non-binding species) and cynomolgus monkeys (binding species) in single- and repeat-dose toxicity studies of up to 6 months duration, respectively. There was no evidence of acute or chronic toxicity identified.
Impairment of Fertility: Reproduction studies have been conducted in cynomolgus monkeys at doses up to 25 times that of the weekly human maintenance dose of 2 mg/kg trastuzumab and have revealed no evidence of impaired fertility.
Teratogenicity: Reproduction studies have been conducted in cynomolgus monkeys at doses up to 25 times that of the weekly human maintenance dose of 2 mg/kg trastuzumab and have revealed no evidence of harm to the foetus. However, when assessing the risk of reproductive toxicity to humans, it is also important to consider the significance of the rodent form of the HER2 receptor in normal embryonic development and the embryonic death in mutant mice lacking this receptor. Placental transfer of trastuzumab during the early (days 20-50 of gestation) and late (days 120-150 of gestation) foetal development period was observed.
Other: Lactation: A study conducted in lactating cynomolgus monkeys at doses 25 times that of the weekly human maintenance dose of 2 mg/kg trastuzumab demonstrated that trastuzumab is secreted in the milk. The presence of trastuzumab in the serum of infant monkeys was not associated with any adverse effects on their growth or development from birth to 1 month of age.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image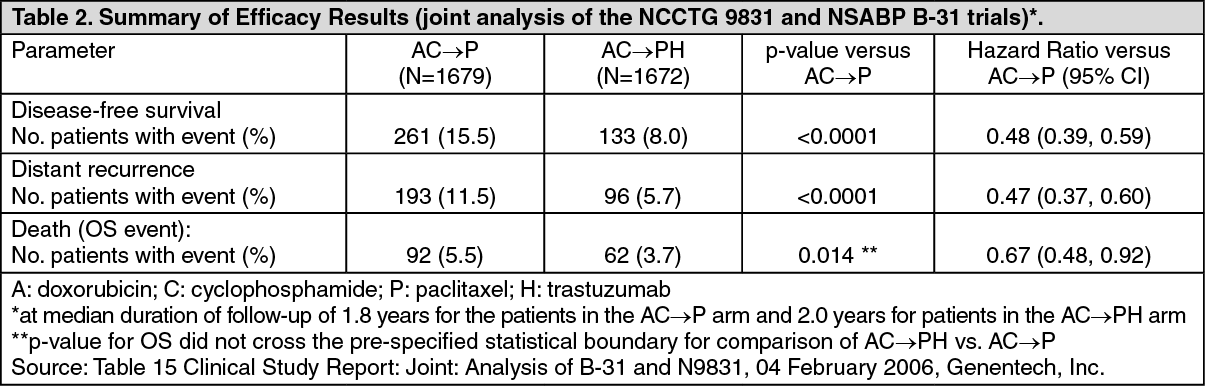 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image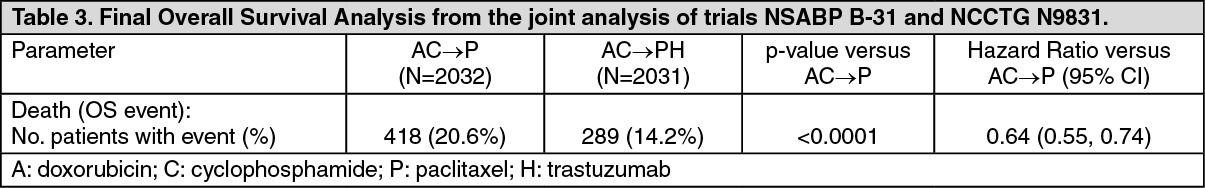 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image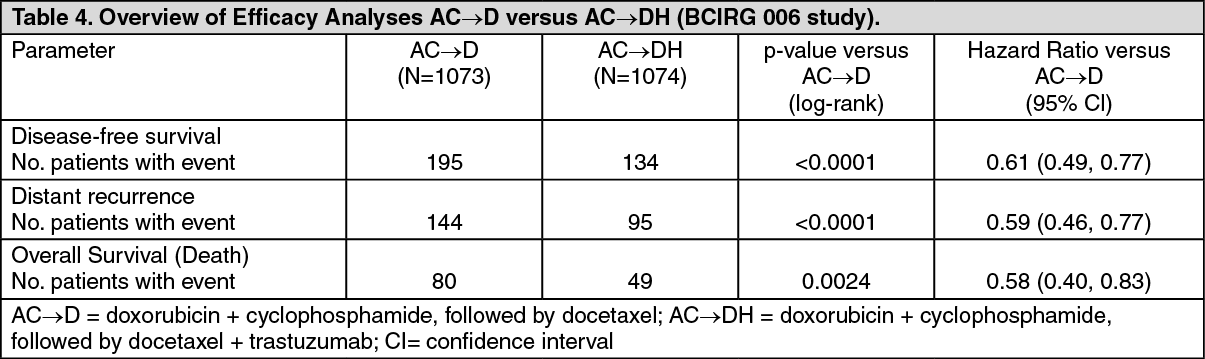 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image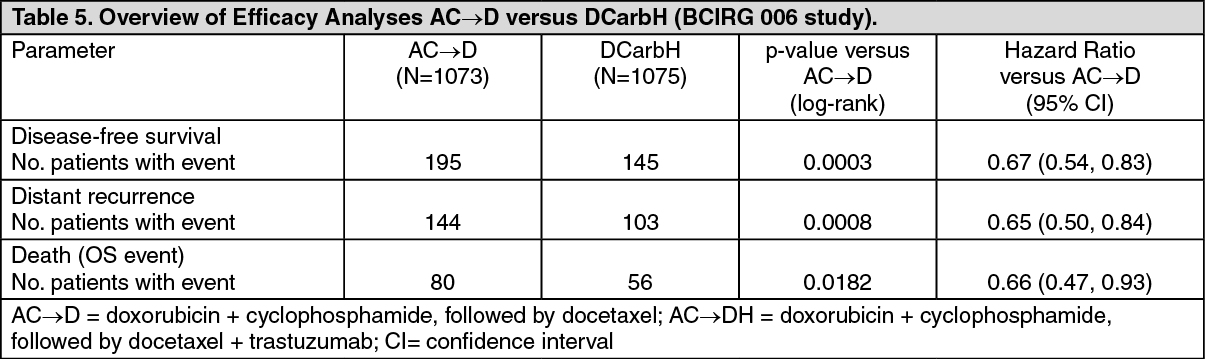 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image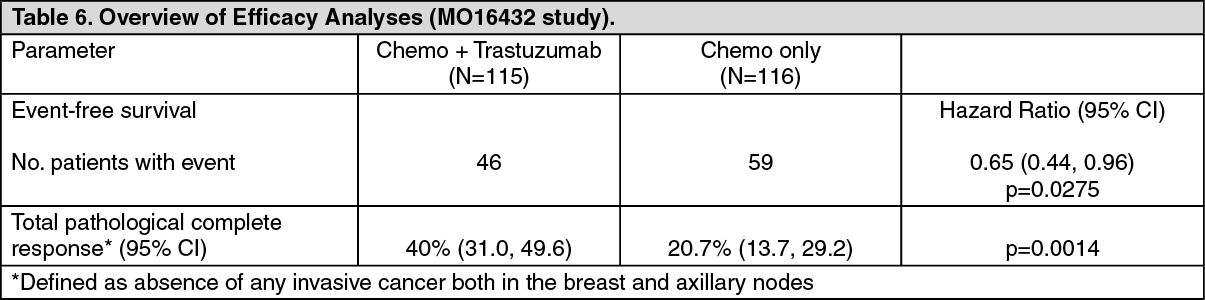 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image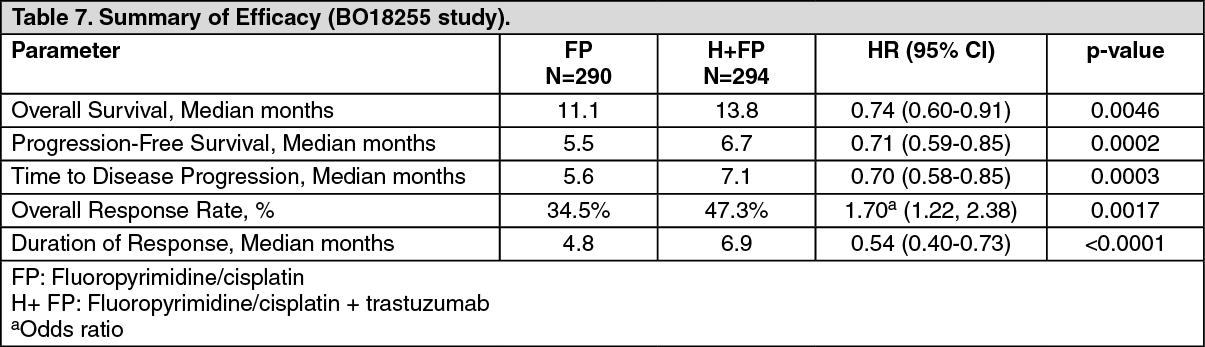 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image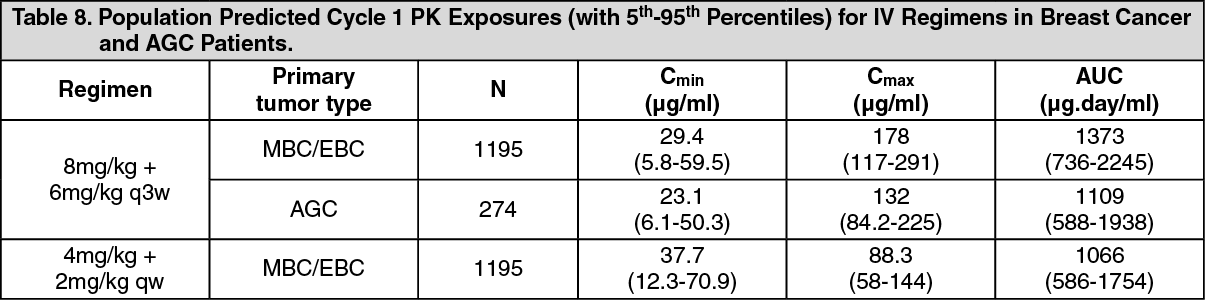 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image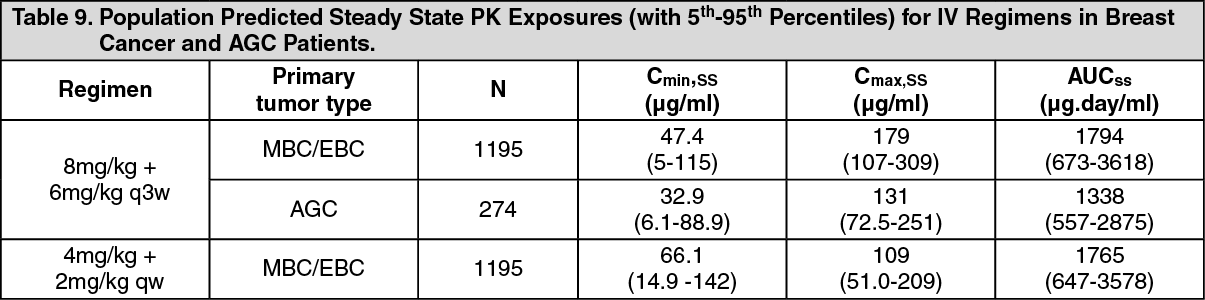 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image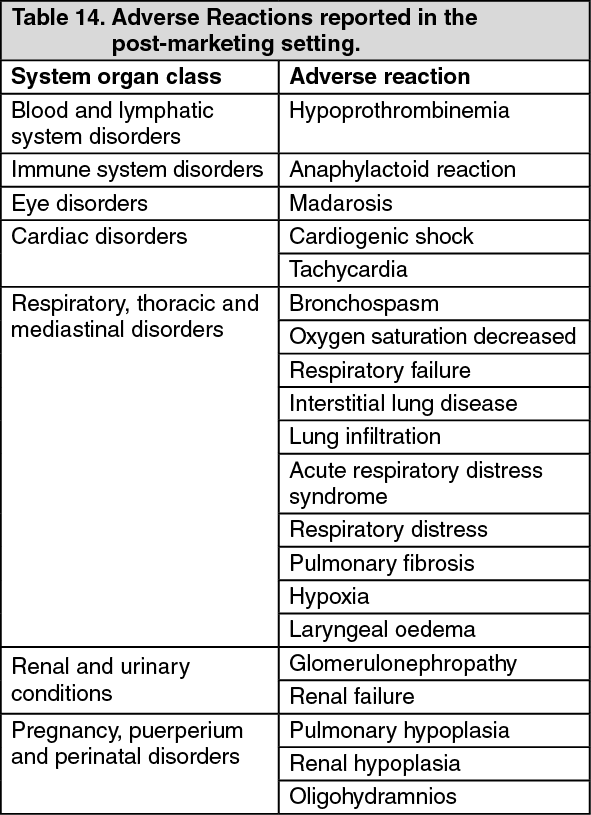 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image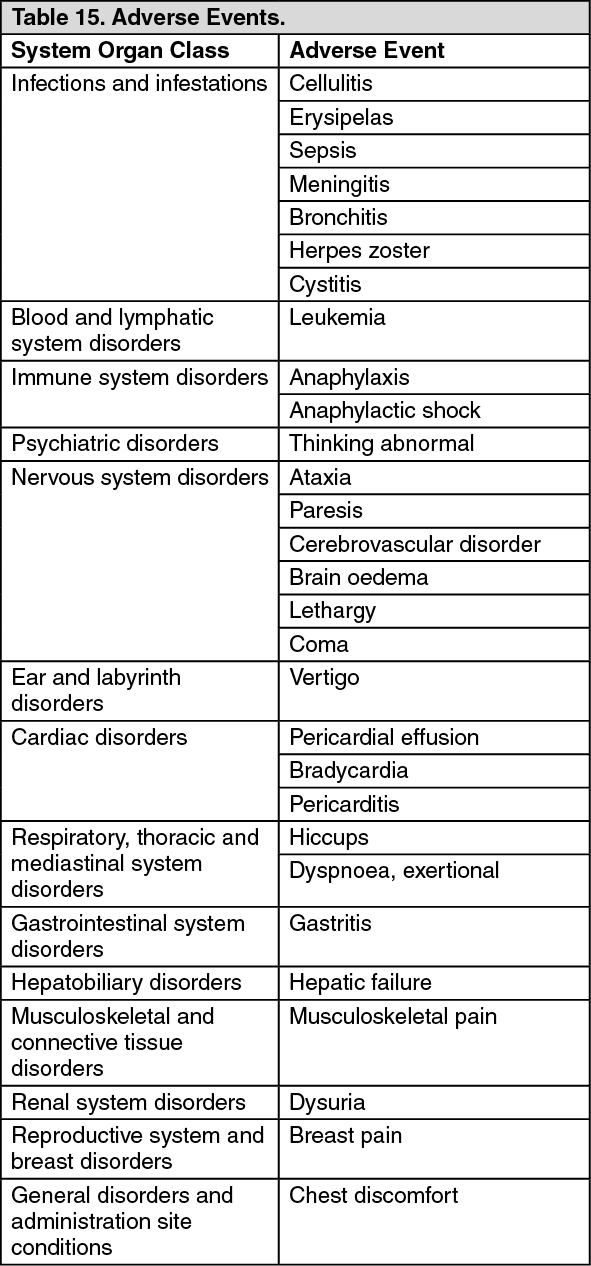 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
