Suicidal Behavior and Ideation: Antiepileptic drugs (AEDs), including INOVELON, increase the risk of suicidal thoughts or behavior in patients taking these drugs for any indication. Patients treated with any AED for any indication should be monitored for the emergence or worsening of depression, suicidal thoughts or behavior, and/or any unusual changes in mood or behavior.
Pooled analyses of 199 placebo-controlled clinical trials (mono- and adjunctive therapy) of 11 different AEDs showed that patients randomized to one of the AEDs had approximately twice the risk (adjusted Relative Risk 1.8, 95% CI:1.2, 2.7) of suicidal thinking or behavior compared to patients randomized to placebo. In these trials, which had a median treatment duration of 12 weeks, the estimated incidence rate of suicidal behavior or ideation among 27,863 AED-treated patients was 0.43%, compared to 0.24% among 16,029 placebo-treated patients, representing an increase of approximately one case of suicidal thinking or behavior for every 530 patients treated. There were four suicides in drug-treated patients in the trials and none in placebo-treated patients, but the number is too small to allow any conclusion about drug effect on suicide.
The increased risk of suicidal thoughts or behavior with AEDs was observed as early as 1 week after starting drug treatment with AEDs and persisted for the duration of treatment assessed. Because most trials included in the analysis did not extend beyond 24 weeks, the risk of suicidal thoughts or behavior beyond 24 weeks could not be assessed.
The risk of suicidal thoughts or behavior was generally consistent among drugs in the data analyzed. The finding of increased risk with AEDs of varying mechanisms of action and across a range of indications suggests that the risk applies to all AEDs used for any indication. The risk did not vary substantially by age (5-100 years) in the clinical trials analyzed. Table 2 shows absolute and relative risk by indication for all evaluated AEDs. (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The relative risk for suicidal thoughts or behavior was higher in clinical trials for epilepsy than in clinical trials for psychiatric or other conditions, but the absolute risk differences were similar for the epilepsy and psychiatric indications.
Anyone considering prescribing INOVELON or any other AED must balance the risk of suicidal thoughts or behavior with the risk of untreated illness. Epilepsy and many other illnesses for which AEDs are prescribed are themselves associated with morbidity and mortality and an increased risk of suicidal thoughts and behavior. Should suicidal thoughts and behavior emerge during treatment, consider whether the emergence of these symptoms in any given patient may be related to the illness being treated.
Patients, their caregivers, and families should be informed that AEDs increase the risk of suicidal thoughts and behavior and should be advised of the need to be alert for the emergence or worsening of the signs and symptoms of depression, any unusual changes in mood or behavior, or the emergence of suicidal thoughts, behavior, or thoughts about self-harm. Behaviors of concern should be reported immediately to healthcare providers.
Central Nervous System Reactions: Use of INOVELON has been associated with central nervous system-related adverse reactions in the controlled clinical trial of patients 4 years or older with Lennox-Gastaut Syndrome. The most significant of these can be classified into two general categories: 1) somnolence or fatigue, and 2) coordination abnormalities, dizziness, gait disturbances, and ataxia.
Somnolence was reported in 24% of INOVELON-treated patients compared to 13% of patients on placebo, and led to study discontinuation in 3% of INOVELON-treated patients compared to 0% of patients on placebo. Fatigue was reported in 10% of INOVELON-treated patients compared to 8% of patients on placebo patients. It led to study discontinuation in 1% of INOVELON-treated patients and 0% of patients on placebo patients.
Dizziness was reported in 2.7% of INOVELON-treated patients compared to 0% of patients on placebo, and did not lead to study discontinuation.
Ataxia and gait disturbance were reported in 5.4% and 1.4% of INOVELON-treated patients, respectively, compared to no patient on placebo. None of these reactions led to study discontinuation.
Accordingly, patients should be advised not to drive or operate machinery until they have gained sufficient experience on INOVELON to gauge whether it adversely affects their ability to drive or operate machinery.
QT Shortening: Formal cardiac ECG studies demonstrated shortening of the QT interval (mean = 20 msec, for doses ≥ 2400 mg twice daily) with INOVELON. In a placebo-controlled study of the QT interval, a higher percentage of INOVELON -treated subjects (46% at 2400 mg, 46% at 3200 mg, and 65% at 4800 mg) had a QT shortening of greater than 20 msec at T
max compared to placebo (5-10%).
Reductions of the QT interval below 300 msec were not observed in the formal QT studies with doses up to 7200 mg per day. Moreover, there was no signal for drug-induced sudden death or ventricular arrhythmias.
The degree of QT shortening induced by INOVELON is without any known clinical risk. Familial Short QT syndrome is associated with an increased risk of sudden death and ventricular arrhythmias, particularly ventricular fibrillation. Such events in this syndrome are believed to occur primarily when the corrected QT interval falls below 300 msec. Non-clinical data also indicate that QT shortening is associated with ventricular fibrillation.
Patients with Familial Short QT syndrome should not be treated with INOVELON. Caution should be used when administering INOVELON with other drugs that shorten the QT interval (see Contraindications).
Multi-organ Hypersensitivity/Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS): Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS), also known as multi-organ hypersensitivity, has been reported in patients taking antiepileptic drugs, including INOVELON. DRESS may be fatal or life-threatening. DRESS typically, although not exclusively, presents with fever, rash, and/or lymphadenopathy, in association with other organ system involvement, such as hepatitis, nephritis, hematological abnormalities, myocarditis, or myositis, sometimes resembling an acute viral infection. Eosinophilia is often present. It is important to note that early manifestations of hypersensitivity, such as fever or lymphadenopathy, may be present even though rash is not evident. Because this disorder is variable in its expression, other organ systems not noted here may be involved.
All cases of DRESS identified in clinical trials with INOVELON occurred in pediatric patients less than 12 years of age, occurred within 4 weeks of treatment initiation, and resolved or improved with INOVELON discontinuation. DRESS has also been reported in adult and pediatric patients taking INOVELON in the postmarketing setting.
If DRESS is suspected, the patient should be evaluated immediately, INOVELON should be discontinued, and alternative treatment should be started.
Withdrawal of AEDs: As with all antiepileptic drugs, INOVELON should be withdrawn gradually to minimize the risk of precipitating seizures, seizure exacerbation, or status epilepticus. If abrupt discontinuation of the drug is medically necessary, the transition to another AED should be made under close medical supervision. In clinical trials, INOVELON discontinuation was achieved by reducing the dose by approximately 25% every 2 days.
Status Epilepticus: Estimates of the incidence of treatment emergent status epilepticus among patients treated with INOVELON are difficult because standard definitions were not employed. In a controlled Lennox-Gastaut Syndrome trial, 3 of 74 (4.1%) INOVELON-treated patients had episodes that could be described as status epilepticus in the INOVELON-treated patients compared with none of the 64 patients in the placebo-treated patients. In all controlled trials that included patients with different epilepsies, 11 of 1240 (0.9%) INOVELON-treated patients had episodes that could be described as status epilepticus compared with none of 635 patients in the placebo-treated patients.
Leukopenia: INOVELON has been shown to reduce white cell count. Leukopenia (white cell count < 3X10
9/L) was more commonly observed in INOVELON-treated patients 43 of 1171 (3.7%) than placebo-treated patients, 7 of 579 (1.2%) in all controlled trials.
Use in Specific Populations: Renal Impairment: Rufinamide pharmacokinetics in patients with severe renal impairment (creatinine clearance < 30 mL/min) was similar to that of healthy subjects. Dose adjustment in patients undergoing dialysis should be considered (see Pharmacology under Actions).
Hepatic Impairment: Use of INOVELON in patients with severe hepatic impairment (Child-Pugh score 10 to 15) is not recommended. Caution should be exercised in treating patients with mild (Child-Pugh score 5 to 6) to moderate (Child-Pugh score 7 to 9) hepatic impairment.
Use in Pregnancy: Pregnancy Category C: There are no adequate and well-controlled studies in pregnant women. INOVELON should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. Rufinamide produced developmental toxicity when administered orally to pregnant animals at clinically relevant doses.
Rufinamide was administered orally to rats at doses of 20, 100, and 300 mg/kg per day and to rabbits at doses of 30, 200, and 1000 mg/kg/day during the period of organogenesis (implantation to closure of the hard palate); the high doses are associated with plasma AUCs ≈2 times the human plasma AUC at the maximum recommended human dose (MRHD, 3200 mg per day). Decreased fetal weights and increased incidences of fetal skeletal abnormalities were observed in rats at doses associated with maternal toxicity. In rabbits, embryo-fetal death, decreased fetal body weights, and increased incidences of fetal visceral and skeletal abnormalities occurred at all but the low dose. The highest dose tested in rabbits was associated with abortion. The no-effect doses for adverse effects on rat and rabbit embryo-fetal development (20 and 30 mg/kg per day, respectively) were associated with plasma AUCs ≈ 0.2 times that in humans at the MRHD.
In a rat pre- and post-natal development study (dosing from implantation through weaning) conducted at oral doses of 5, 30, and 150 mg/kg per day (associated with plasma AUCs up to ≈1.5 times that in humans at the MRHD), decreased offspring growth and survival were observed at all doses tested. A no-effect dose for adverse effects on pre- and post-natal development was not established. The lowest dose tested was associated with plasma AUC < 0.1 times that in humans at the MRHD.
Use in Lactation: Rufinamide is likely to be excreted in human milk. Because of the potential for serious adverse reactions in nursing infants from INOVELON, a decision should be made whether to discontinue nursing or discontinue the drug taking into account the importance of the drug to the mother.
Use in Children: Safety and effectiveness have been established in pediatric patients 1 to 7 years of age. The effectiveness of INOVELON in pediatric patients 4 years of age and older was based upon an adequate and well-controlled trial of INOVELON that included both adults and pediatric patients, 4 years of age and older, with Lennox Gastaut Syndrome. The effectiveness in patients in patients 1 to less than 4 years was based upon a bridging pharmacokinetic and safety study (see Dosage & Administration, Adverse Reactions, and Pharmacology: Pharmacodynamics: Clinical Studies under Actions). The pharmacokinetics of rufinamide in the pediatric patients, ages 1 to less than 4 years of age is similar to children older than 4 years of age and adults (see Pharmacology under Actions).
Safety and effectiveness in pediatric patients below the age of 1 year has not been established.
Use in Elderly: Clinical studies of INOVELON did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Pharmacokinetics of rufinamide in the elderly are similar to that in the young subjects (see Pharmacology under Actions).


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image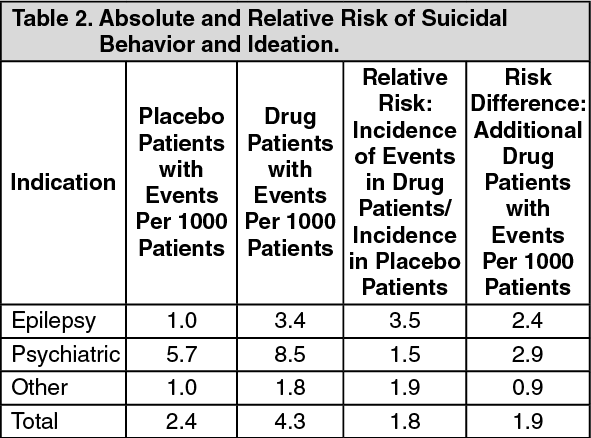 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image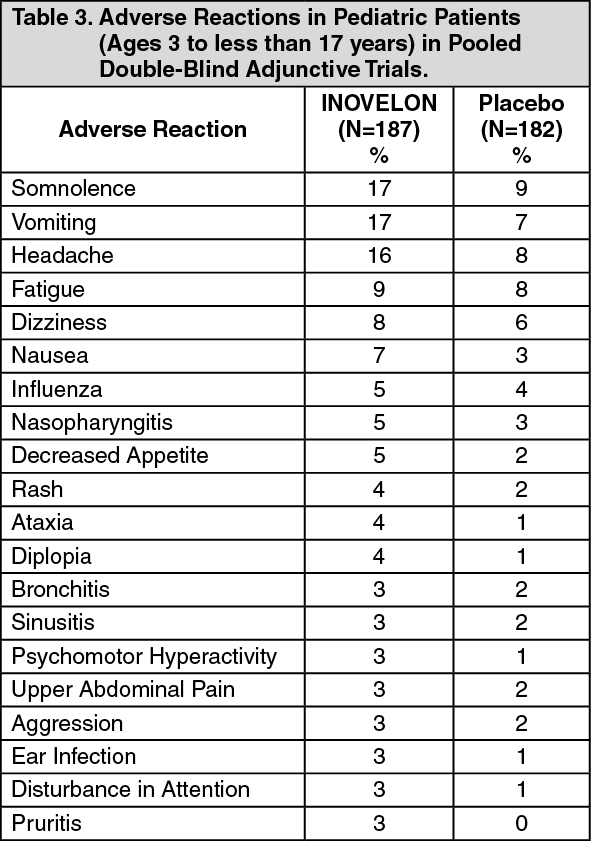 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image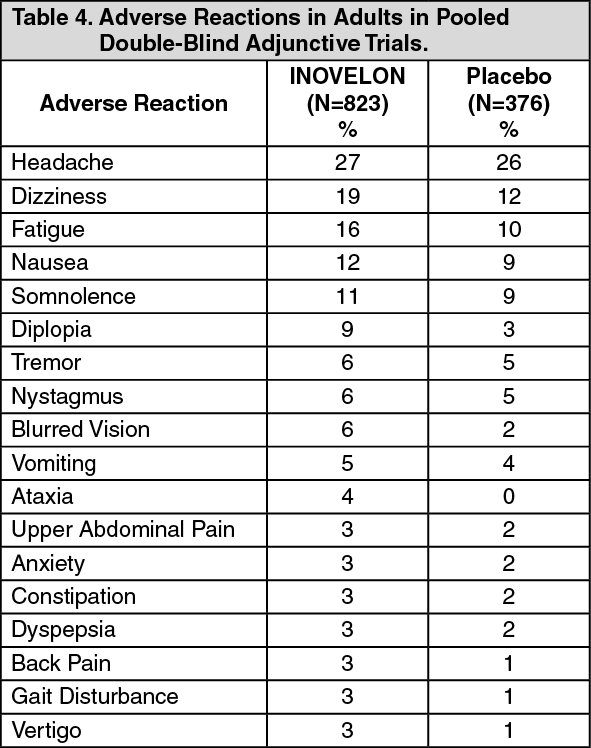 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image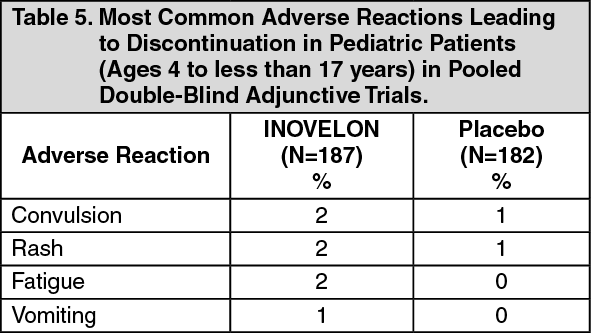 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image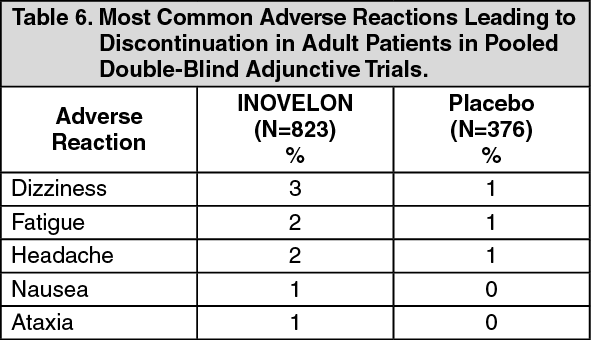 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image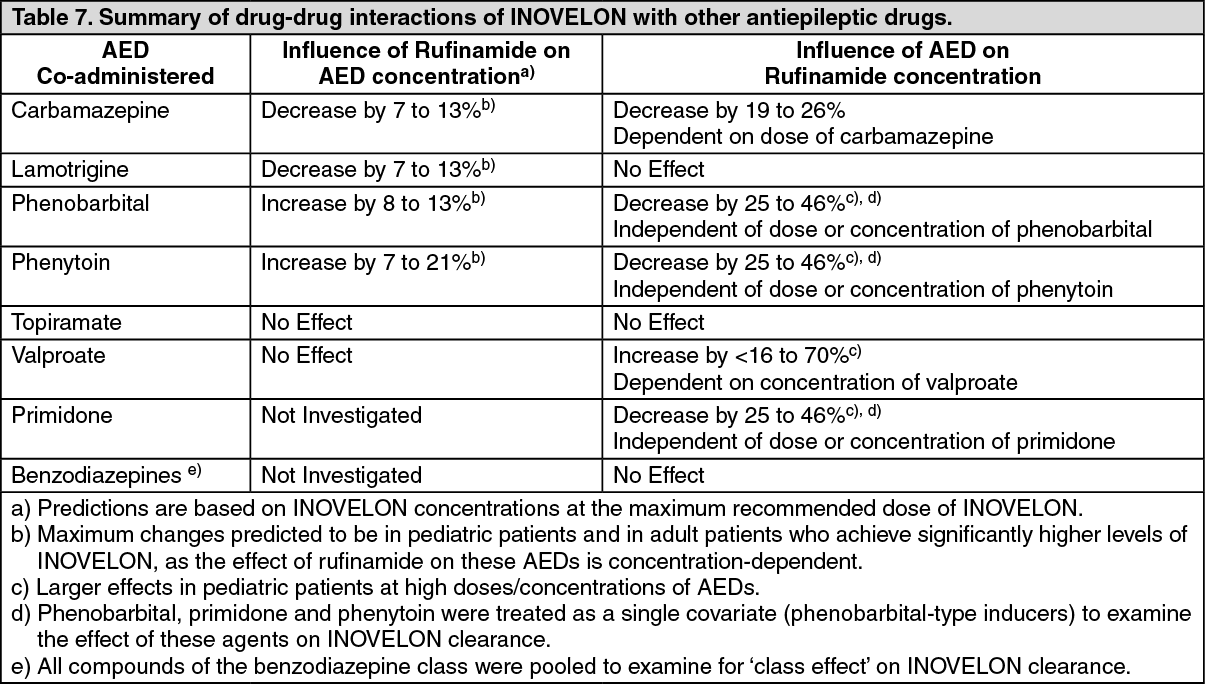 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out