Pharmacology: Pharmacodynamics: Mechanism of Action: Paclitaxel is a novel antimicrotubule agent that promotes the assembly of microtubules from tubulin dimers. It stabilizes microtubules by preventing depolymerization resulting in the inhibition of the normal dynamic reorganization of the microtubule network essential for cellular functions. Paclitaxel also induces abnormal arrays or "bundles" of microtubules throughout the cell cycle and multiple asters of microtubules during mitosis.
Clinical Studies: Ovarian Cancer: Four trials of Paclitaxel as single agent in patients who had received prior platinum based first line treatment, confirmed its efficacy as salvage therapy for ovarian carcinoma. These trials, which enrolled a total of 177 patients, were conducted at John Hopkins Oncology Centre, Albert Einstein Cancer Centre, GOG and NCI, used a dose ranging between 170-250 mg/m
2. Out of the 157 patients evaluable, there were 55 responders giving an overall response rate of 35% with 16 CRs (10%) and 39 PRs (25%). Out of the 52 patients categorized as clinically platinum resistant, 15 (29%) responded. These four trials led two significant conclusions. First, Paclitaxel is clearly active against ovarian carcinoma previously treated with platinum based chemotherapy. Second, the level of activity does not appear to be influenced by the results or prior platinum based therapy.
In yet another study at NCI a total of 1819 patients were enrolled to be treated with Paclitaxel 135 mg/m
2 over 24 hrs every 3 weeks. A report on the first 1000 patients noted 935 evaluable patients, 650 of whom had clinically measurable disease. Among these 23 CRs (4%) and 118 PRs (18%) were observed for an overall response rate of 22%.
Metastatic Breast Cancer: After the observation of significant antitumour activity of Paclitaxel in women with stage IV breast cancer (minimally pretreated), a phase II study was initiated in 33 women with anthracycline resistance breast cancer, at those of Paclitaxel 175 mg/m
2 in a 3 h infusion every 3 weeks. There were 2 CRs (6%) and 12 PRs (36%) giving an overall response rate of 42%, median time to progression was 24 weeks and median survival was 41 weeks. Another phase II study conducted at MSKCC yielded 28% response rate in 51 patients who had received a median of 3 prior chemotherapeutic regiments for metastatic disease.
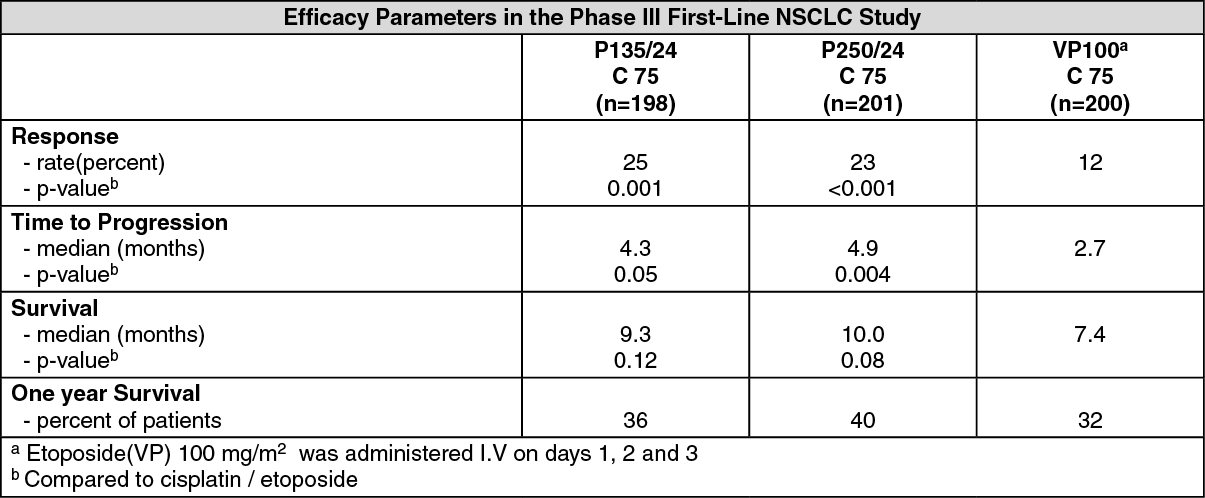
Non-small cell lung cancer: In a Phase III open label randomized study conducted by the ECOG, 599 patients were randomized to either Paclitaxel (P) 135 mg/m
2 as a 24-hour infusion in combination with cisplatin (c) 75 mg/m
2, Paclitaxel (P) 250 mg/m
2 as a 24-hour infusion in combination with cisplatin (c) 75 mg/m
2 with G-CSF support, or cisplatin (c) 75 mg/m
2 on day 1, followed by etoposide (VP) 100 mg/m
2 on days 1, 2 and 3 (control).
Response rates, median time to progression, median survival, and one year survival rates are given in the following table. The reported p-values have not been adjusted for multiple comparisons. There were statistically significant differences favoring each of the Paclitaxel plus cisplatin arms for response rate and time to tumor progression. There was no statistically significant difference in survival between Paclitaxel plus cisplatin arm and the cisplatin plus etoposide arm. See table.
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
In the ECOG study, the Functional Assessment of Cancer Therapy-Lung (FACT-L) questionnaire had seven subscales that measured subjective assessment of treatment. Of the seven, the Lung Cancer Specific Symptoms subscale favored the paclitaxel 135 mg/m
2/24 hour plus cisplatin arm compared to the cisplatin/etoposide arm. For all other factors, there was no difference in the treatment groups.
Pharmacokinetics: Absorption: With a 24-hour infusion, a 30% increase in dose increased the C
max by 87% with a 3-hour infusion, a 30% dose increase resulted in a 68% increase in the C
max.
Distribution: The mean steady state volume of distribution has range from 227 to 688 L/m
2, indicating extensive extravascular distribution and/or tissue binding. 89% to 98% of drug is bound to plasma proteins.
Metabolism: In vitro studies with human liver microsomes and tissue slices showed that paclitaxel was metabolized primarily to 6α, 3'-
p-hydroxypaclitaxel the cytochrome P450 isozyme CYP2C8; and to two minor metabolites, 3-
p-hydroxypaclitaxel and 6α, 3'-
p-hydroxypaclitaxel by CYP3A4. In vitro, the metabolism of paclitaxel to 6α-hydroxypaclitaxel was inhibited by a number of agents such as ketoconazole, verapamil, diazepam, quinidine, dexamethasone, cyclosporine, teniposide, etoposide and vincristine.
Excretion: Following intravenous administration, paclitaxel exhibits a biphasic decline in plasma concentrations. The initial rapid decline represents distributions to the peripheral compartment and elimination of the drug. The later phase is due, in part, to a relatively slow efflux of paclitaxel from the peripheral compartment. In patients treated with doses of 135 and 175 mg/m
2 given as 3 and 24 hour infusions, mean terminal half-life has ranged from 13.1 to 52.7 hours and total body clearance has ranged from 12.2 to 23.8 L/h/m
2.
Elimination: After intravenous administration of 15-275 mg/m
2 doses of Paclitaxel as 1, 6 or 24-hour infusions, mean values for cumulative urinary recovery unchanged drug ranged from 1.3% to 12.6% of the dose. This indicates extensive non-renal clearance of paclitaxel.
CLINICAL PHARMACOLOGY: The pharmacokinetics of Paclitaxel has been evaluated in adult cancer patients who received single doses of 15-135 mg/m
2 given by 1-hour infusion (n=15), 30-275 mg/m
2 given by 6-hour infusions (n=36), and 135-275 mg/m
2 given by 24 hour infusions (n=54). Following intravenous administration of Paclitaxel, the drug exhibited a biphasic decline in plasma concentrations. The initial rapid decline represents distribution to the peripheral compartment and significant elimination of the drug. The later phase was due, in part, to a relatively slow efflux of Paclitaxel from the peripheral compartment. Values for mean terminal phase half-life; total body clearance and apparent volume of distribution at steady state were determined following 1 hour and 6 hour infusions at dosing levels of 15-275 mg/m
2. Mean (standard deviation) terminal half-life was estimated to range from 5.3 (4.6) to 17.4 (4.7) hours. Mean (SD) values for total body clearance ranged from 5.8 (2.3) to 16.3 (2.3) l/h/m
2. The mean (SD) steady state volume of distribution ranged from 42 (15) to 162 (133) l/m
2, indicating extravascular distribution and/or tissue binding of Paclitaxel.
In vitro studies of binding to human serum proteins, using Paclitaxel concentrations ranging from 0.1 to 50 micrograms/ml, indicated that between 89-98% of the drug is bound; the presence of cimetidine, ranitidine or dexamethasone did not effect the protein binding of Paclitaxel. Mean (SD) C
max values ranged from 435 (111) to 802 (260) ng/ml following 24 hour infusions at doses of Paclitaxel of 200 to 275 mg/m
2, and were approximately 10-30% of those following 6 hour infusions of equivalent doses. After administration of doses of Paclitaxel of 170 mg/m
2 or higher by infusion lasting 6 or 24 hours, plasma concentrations above 85 ng/ml, the level shown to be pharmacologically active in vitro, were regularly observed for at least 6 to 12 hours. The disposition of Paclitaxel has not been fully elucidated in humans. After intravenous administration of 15-175 mg/m
2 doses of Paclitaxel as 1, 6, 24 hour infusions, mean (SD) values for cumulative urinary recovery of unchanged drug ranged from 1.3% (0.5%) to 12.6% (16.2%) of the dose, indicating extensive non-renal clearance. Paclitaxel has been shown to be metabolized in the liver in animals and there is evidence suggesting hepatic metabolism in human. High Paclitaxel concentrations have been reported in the bile of patients treated with the drug. The effect of renal of hepatic dysfunction on the disposition of Paclitaxel has not been investigated. Possible interactions of Paclitaxel with concomitantly administered medications have been formally investigated.
Toxicology: Preclinical Safety Data: Carcinogenesis, Mutagenesis, Impairment of Fertility: The carcinogenic potential of Paclitaxel has not been studied. Paclitaxel has been shown to be mutagenic in vitro (chromosome aberrations in human lymphocytes) and in vivo (micronucleus test in mice) mammalian test systems, however, it did not induce mutagenicity in the Ames test or the CHO/HGPRT gene mutation assay. Paclitaxel at an I.V. dose of 1 mg/kg (6 mg/m
2) produced low fertility and fetal toxicity in rats. Paclitaxel has also been shown to be maternal and embryo-fetal toxic in rabbits receiving the drug at an I.V. dose of 3 mg/kg (33 mg/m
2) during organogenesis. (See Warnings.)


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
