Pharmacotherapeutic group: Other antiepileptics.
ATC code: N03AX09.
Pharmacology: Pharmacodynamics: Mechanism of action: The results of pharmacological studies suggest that lamotrigine is a use- and voltage-dependent blocker of voltage gated sodium channels. It inhibits sustained repetitive firing of neurones and inhibits release of glutamate (the neurotransmitter which plays a key role in the generation of epileptic seizures). These effects are likely to contribute to the anticonvulsant properties of lamotrigine.
In contrast, the mechanisms by which lamotrigine exerts its therapeutic action in bipolar disorder have not been established, although interaction with voltage gated sodium channels is likely to be important.
Pharmacodynamic effects: In tests designed to evaluate the central nervous system effects of medicinal products, the results obtained using doses of 240 mg lamotrigine administered to healthy volunteers did not differ from placebo, whereas both 1000 mg phenytoin and 10 mg diazepam each significantly impaired fine visual motor coordination and eye movements, increased body sway and produced subjective sedative effects.
In another study, single oral doses of 600mg carbamazepine significantly impaired fine visual motor coordination and eye movements, while increasing both body sway and heart rate, whereas results with lamotrigine at doses of 150mg and 300mg did not differ from placebo.
Clinical efficacy and safety in children aged 1 to 24 months: The efficacy and safety of adjunctive therapy in partial seizures in patients aged 1 to 24 months has been evaluated in a small double-blind placebo-controlled withdrawal study. Treatment was initiated in 177 subjects, with a dose titration schedule similar to that of children aged 2 to 12 years. Lamotrigine 2 mg tablets are the lowest strength available, therefore the standard dosing schedule was adapted in some cases during the titration phase (for example, by administering a 2 mg tablet on alternate days when the calculated dose was less than 2 mg). Serum levels were measured at the end of week 2 of titration and the subsequent dose either reduced or not increased if the concentration exceeded 0.41 µg/mL, the expected concentration in adults at this time point. Dose reductions of up to 90% were required in some patients at the end of week 2. Thirty-eight responders (> 40% decrease in seizure frequency) were randomised to placebo or continuation of lamotrigine. The proportion of subjects with treatment failure was 84% (16/19 subjects) in the placebo arm and 58% (11/19 subjects) in the lamotrigine arm. The difference was not statistically significant: 26.3%, CI95% -2.6% <> 50.2%, p=0.07.
A total of 256 subjects between 1 to 24 months of age have been exposed to lamotrigine in the dose range 1 to 15 mg/kg/day for up to 72 weeks. The safety profile of lamotrigine in children aged 1 month to 2 years was similar to that in older children except that clinically significant worsening of seizures (>=50%) was reported more often in children under 2 years of age (26%) as compared to older children (14%).
Clinical efficacy and safety in Lennox-Gastaut syndrome: There are no data for monotherapy in seizures associated with Lennox-Gastaut syndrome.
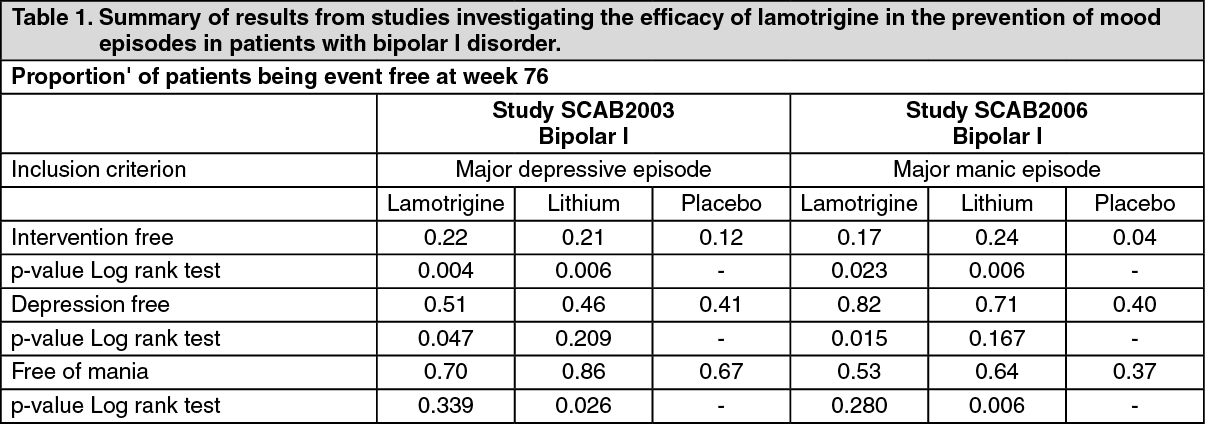
Clinical efficacy in the prevention of mood episodes in patients with bipolar disorder: The efficacy of lamotrigine in the prevention of mood episodes in patients with bipolar I disorder has been evaluated in two studies.
Study SCAB2003 was a multicentre, double-blind, double dummy, placebo and lithium-controlled, randomised fixed dose evaluation of the long-term prevention of relapse and recurrence of depression and/or mania in patients with bipolar I disorder who had recently or were currently experiencing a major depressive episode. Once stabilised using lamotrigine monotherapy or adjunctive therapy, patients were randomly assigned into one of five treatment groups: lamotrigine (50, 200, 400 mg/day), lithium (serum levels of 0.8 to 1.1 mMol/L) or placebo for a maximum of 76 weeks (18 months). The primary endpoint was "Time to Intervention for a Mood Episode (TIME)", where the interventions were additional pharmacotherapy or electroconvulsive therapy (ECT).
Study SCAB2006 had a similar design as study SCAB2003, but differed from study SCAB2003 in evaluating a flexible dose of lamotrigine (100 to 400 mg/day) and including patients with bipolar I disorder who had recently or were currently experiencing a manic episode. The results are shown in Table 1. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
In supportive analyses of time to first depressive episode and time to first manic/hypomanic or mixed episode, the lamotrigine-treated patients had significantly longer times to first depressive episode than placebo patients, and the treatment difference with respect to time to manic/hypomanic or mixed episodes was not statistically significant.
The efficacy of lamotrigine in combination with mood stabilisers has not been adequately studied.
Children (10-12 years of age) and Adolescents (13-17 years of age): A multicentre, parallel group, placebo-controlled, double-blind, randomised withdrawal study, evaluated the efficacy and safety of lamotrigine IR as add on maintenance therapy to delay mood episodes in male and female children and adolescents (age 10-17 years) who had been diagnosed with bipolar I disorder and who had remitted or improved from a bipolar episode while treated with lamotrigine in combinations with concomitant antipsychotic or other mood stabilising drugs. The result of the primary efficacy analysis (time to occurrence of a bipolar event - TOBE) did not reach statistical significance (p=0.0717), thus efficacy was not shown. In addition, safety results showed increased reporting of suicidal behaviours in lamotrigine treated patients: 5% (4 patients) in the lamotrigine arm compared to 0 in placebo (see Dosage & Administration).
Study of the effect of lamotrigine on cardiac conduction: A study in healthy adult volunteers evaluated the effect of repeat doses of lamotrigine (up to 400 mg/day) on cardiac conduction, as assessed by 12-lead ECG. There was no clinically significant effect of lamotrigine on QT interval compared to placebo.
Pharmacokinetics: Absorption: Lamotrigine is rapidly and completely absorbed from the gut with no significant first pass metabolism. Peak plasma concentrations occur approximately 2.5 hours after oral administration of lamotrigine. Time to maximum concentration is slightly delayed after food but the extent of absorption is unaffected. There is considerable inter-individual variation in steady state maximum concentrations but within an individual, concentrations rarely vary.
Distribution: Binding to plasma proteins is about 55%. It is very unlikely that displacement from plasma proteins would result in toxicity. The volume of distribution is 0.92 to 1.22 L/kg.
Biotransformation: UDP-glucuronyl transferases have been identified as the enzymes responsible for metabolism of lamotrigine.
Lamotrigine induces its own metabolism to a modest extent depending on dose. However, there is no evidence that lamotrigine affects the pharmacokinetics of other AEDs and data suggest that interactions between lamotrigine and medicinal products metabolised by cytochrome P450 enzymes are unlikely to occur.
Elimination: The apparent plasma clearance in healthy subjects is approximately 30 mL/min. Clearance of lamotrigine is primarily metabolic with subsequent elimination of glucuronide-conjugated material in urine. Less than 10% is excreted unchanged in the urine. Only about 2% of lamotrigine-related material is excreted in faeces. Clearance and half-life are independent of dose. The apparent plasma half-life in healthy subjects is estimated to be approximately 33 hours (range 14 to 103 hours). In a study of subjects with Gilbert's Syndrome, mean apparent clearance was reduced by 32% compared with normal controls but the values are within the range for the general population.
The half-life of lamotrigine is greatly affected by concomitant medicinal products. Mean half-life is reduced to approximately 14 hours when given with glucuronidation-inducing medicinal products such as carbamazepine and phenytoin and is increased to a mean of approximately 70 hours when co-administered with valproate alone (see Dosage & Administration).
Linearity: The pharmacokinetics of lamotrigine are linear up to 450 mg, the highest single dose tested.
Special patient populations: Children: Clearance adjusted for body weight is higher in children than in adults with the highest values in children under five years. The half-life of lamotrigine is generally shorter in children than in adults with a mean value of approximately 7 hours when given with enzyme-inducing medicinal products such as carbamazepine and phenytoin and increasing to mean values of 45 to 50 hours when co-administered with valproate alone (see Dosage & Administration).
Infants aged 2 to 26 months: In 143 paediatric patients aged 2 to 26 months, weighing 3 to 16 kg, clearance was reduced compared to older children with the same body weight, receiving similar oral doses per kg body weight as children older than 2 years. The mean half-life was estimated at 23 hours in infants younger than 26 months on enzyme-inducing therapy, 136 hours when co-administered with valproate and 38 hours in subjects treated without enzyme inducers/inhibitors. The inter-individual variability for oral clearance was high in the group of paediatric patients of 2 to 26 months (47%). The predicted serum concentration levels in children of 2 to 26 months were in general in the same range as those in older children, though higher Cmax levels are likely to be observed in some children with a body weight below 10 kg.
Elderly: Results of a population pharmacokinetic analysis including both young and elderly patients with epilepsy, enrolled in the same trials, indicated that the clearance of lamotrigine did not change to a clinically relevant extent. After single doses apparent clearance decreased by 12% from 35 mL/min at age 20 to 31 mL/min at 70 years. The decrease after 48 weeks of treatment was 10% from 41 to 37 mL/min between the young and elderly groups. In addition, pharmacokinetics of lamotrigine was studied in 12 healthy elderly subjects following a 150 mg single dose. The mean clearance in the elderly (0.39 mL/min/kg) lies within the range of the mean clearance values (0.31 to 0.65 mL/min/kg) obtained in nine studies with non-elderly adults after single doses of 30 to 450 mg.
Renal impairment: Twelve volunteers with chronic renal failure, and another six individuals undergoing hemodialysis were each given a single 100 mg dose of lamotrigine. Mean clearances were 0.42 mL/min/kg (chronic renal failure), 0.33 mL/min/kg (between hemodialysis) and 1.57 mL/min/kg (during hemodialysis), compared with 0.58 mL/min/kg in healthy volunteers. Mean plasma half-lives were 42.9 hours (chronic renal failure), 57.4 hours (between hemodialysis) and 13.0 hours (during hemodialysis), compared with 26.2 hours in healthy volunteers. On average, approximately 20% (range = 5.6 to 35.1) of the amount of lamotrigine present in the body was eliminated during a 4-hour hemodialysis session. For this patient population, initial doses of lamotrigine should be based on the patient's concomitant medicinal products; reduced maintenance doses may be effective for patients with significant renal functional impairment (see Dosage & Administration and Precautions).
Hepatic impairment: A single dose pharmacokinetic study was performed in 24 subjects with various degrees of hepatic impairment and 12 healthy subjects as controls. The median apparent clearance of lamotrigine was 0.31, 0.24 or 0.10 mL/min/kg in patients with Grade A, B, or C (Child-Pugh Classification) hepatic impairment, respectively, compared with 0.34 mL/min/kg in the healthy controls. Initial, escalation and maintenance doses should generally be reduced in patients with moderate or severe hepatic impairment (see Dosage & Administration).
Toxicology: Preclinical safety data: Non clinical data reveal no special hazard for humans based on studies of safety pharmacology, repeated dose toxicity, genotoxicity and carcinogenic potential.
In reproductive and developmental toxicity studies in rodents and rabbits, no teratogenic effects but reduced foetal weight and retarded skeletal ossification were observed, at exposure levels below or similar to the expected clinical exposure. Since higher exposure levels could not be tested in animals due to the severity of maternal toxicity, the teratogenic potential of lamotrigine has not been characterised above clinical exposure.
In rats, enhanced foetal as well as post natal mortality was observed when lamotrigine was administered during late gestation and through the early post natal period. These effects were observed at the expected clinical exposure.
Neurobehavioural effects (a longer latency period for open field exploration, lower frequency of rearing and increased completion time in a swimming maze test) were observed in the offspring of pregnant rats exposed to clinically relevant exposures of lamotrigine during organogenesis.
In juvenile rats, an effect on learning in the Biel maze test, a slight delay in balanopreputial separation and vaginal patency and a decreased postnatal body weight gain in F1 animals were observed at exposures approximately two times higher than the therapeutic exposures in human adults.
Animal experiments did not reveal impairment of fertility by lamotrigine. Lamotrigine reduced foetal folic acid levels in rats. Folic acid deficiency is assumed to be associated with an enhanced risk of congenital malformations in animals as well as in humans. Lamotrigine caused a dose-related inhibition of the hERG channel tail current in human embryonic kidney cells. The IC50 was approximately nine times above the maximum therapeutic free concentration. Lamotrigine did not cause QT prolongation in animals at exposures up to approximately two times the maximum therapeutic free concentration. In a clinical study, there was no clinically significant effect of lamotrigine on QT interval in healthy adult volunteers (see Pharmacodynamics as previously mentioned).


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image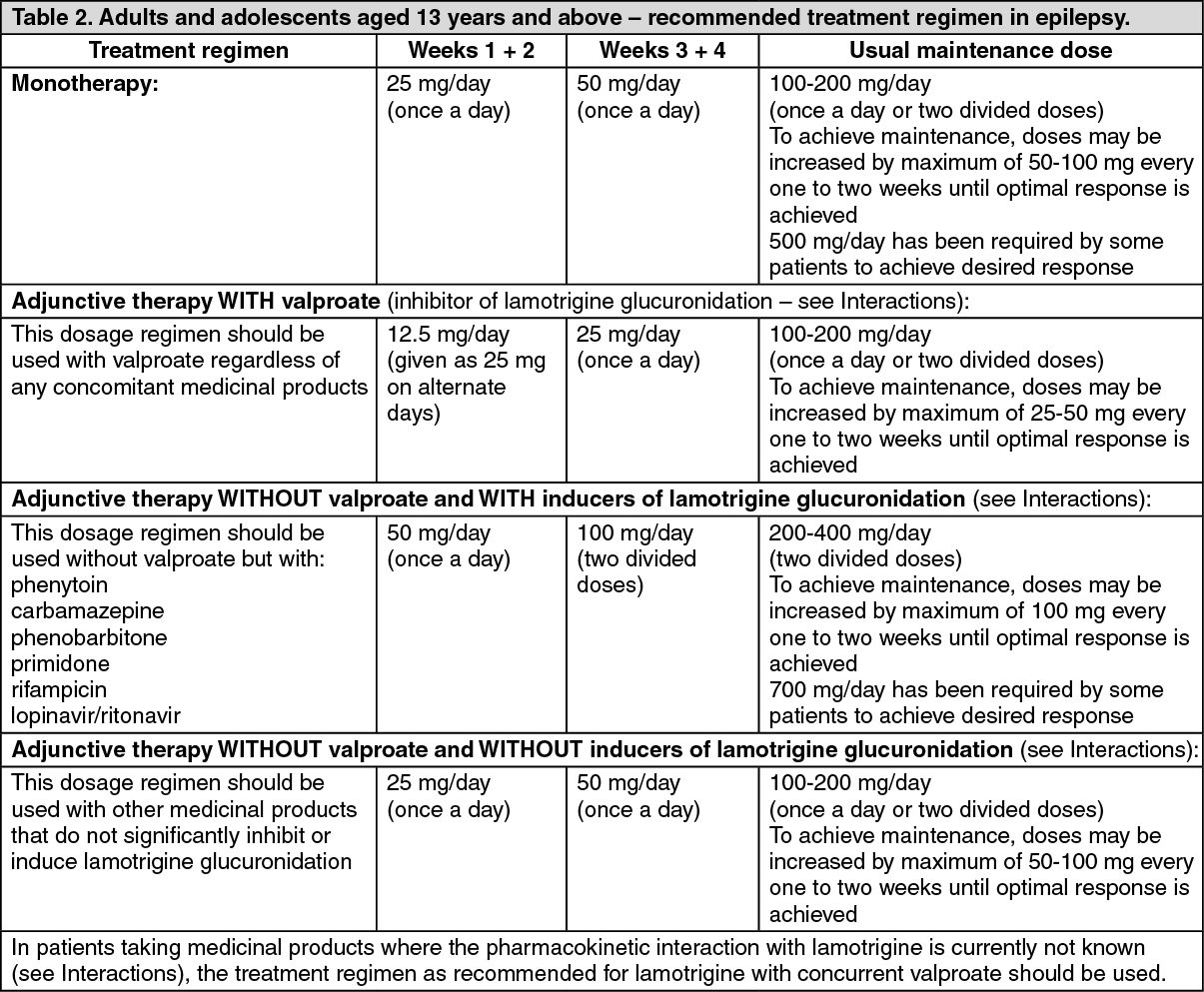 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image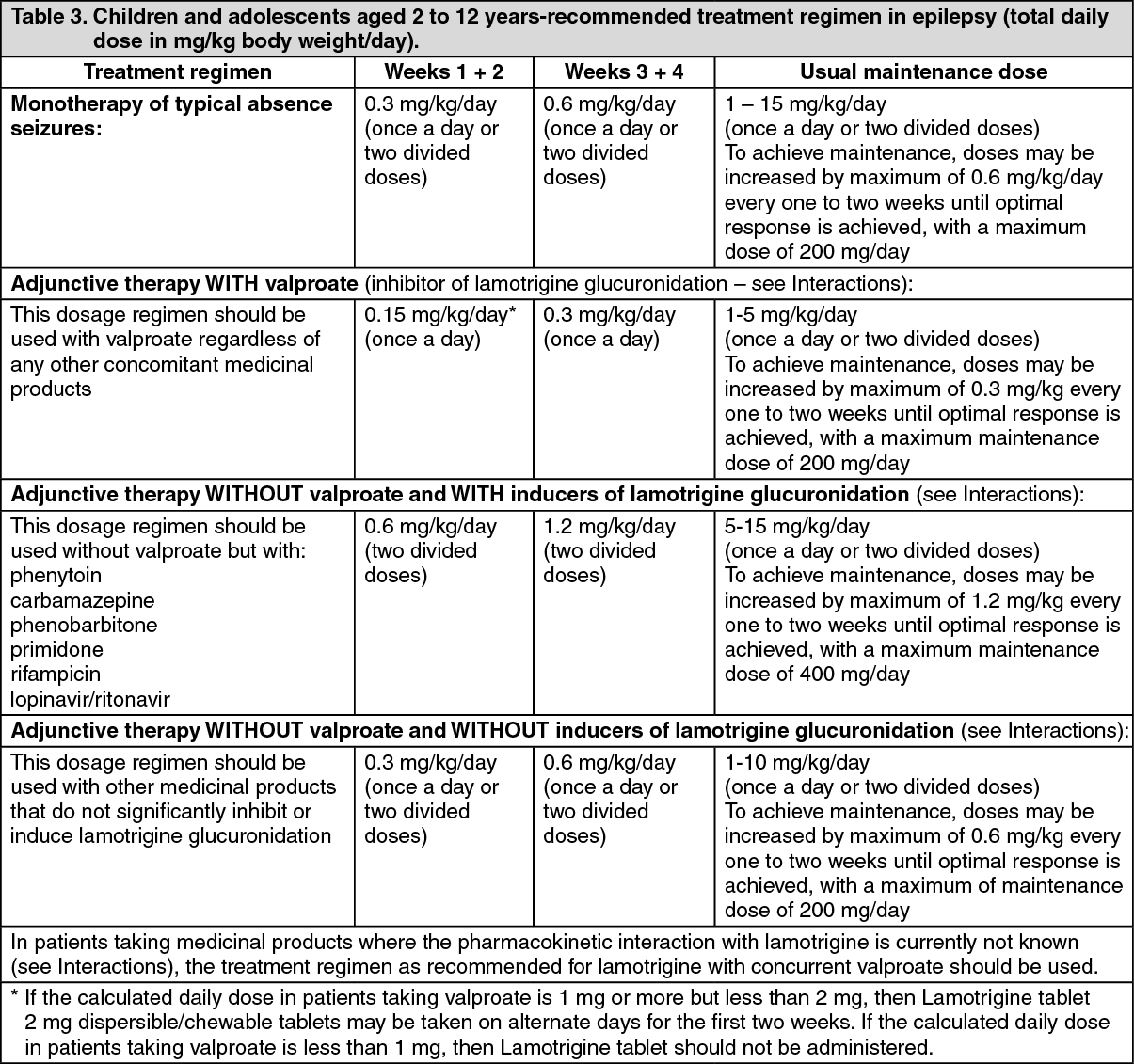 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image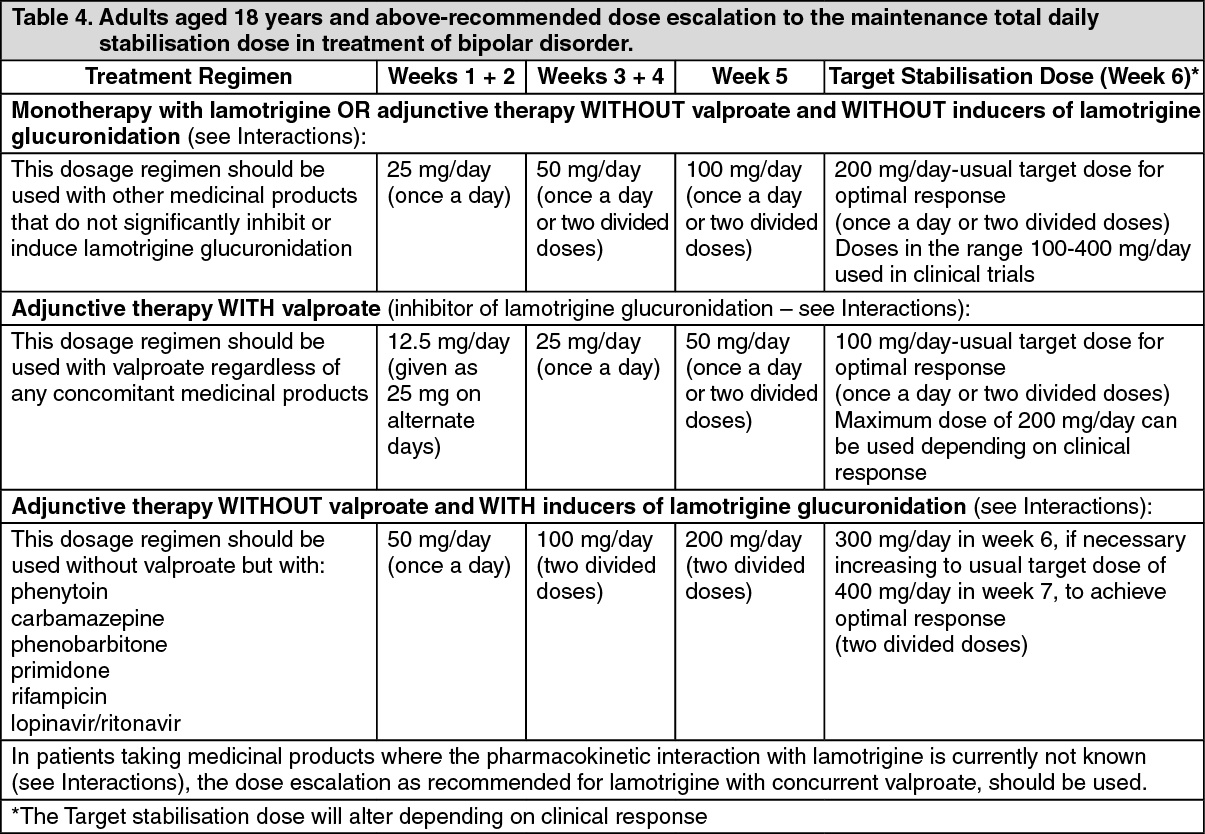 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image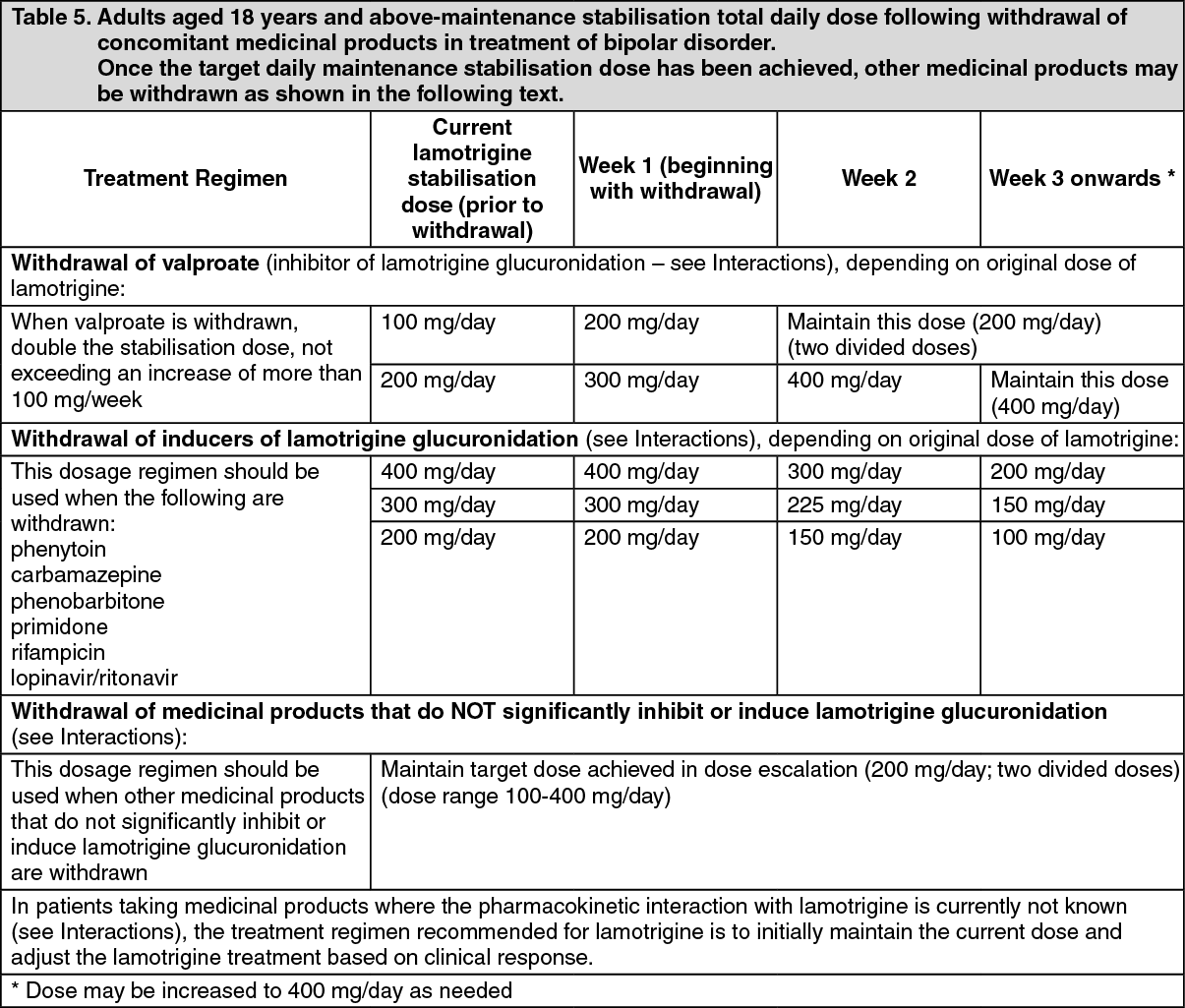 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image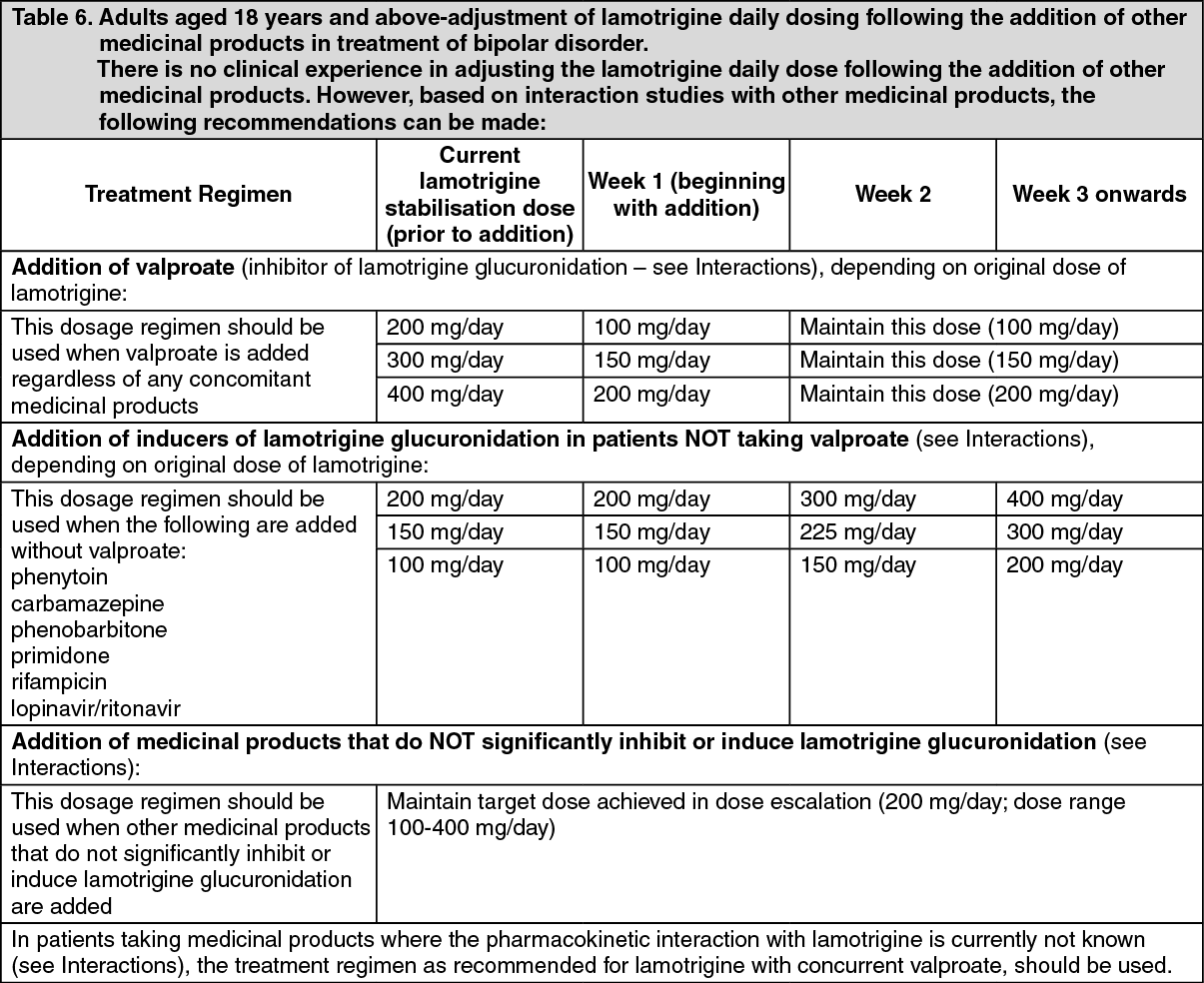 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image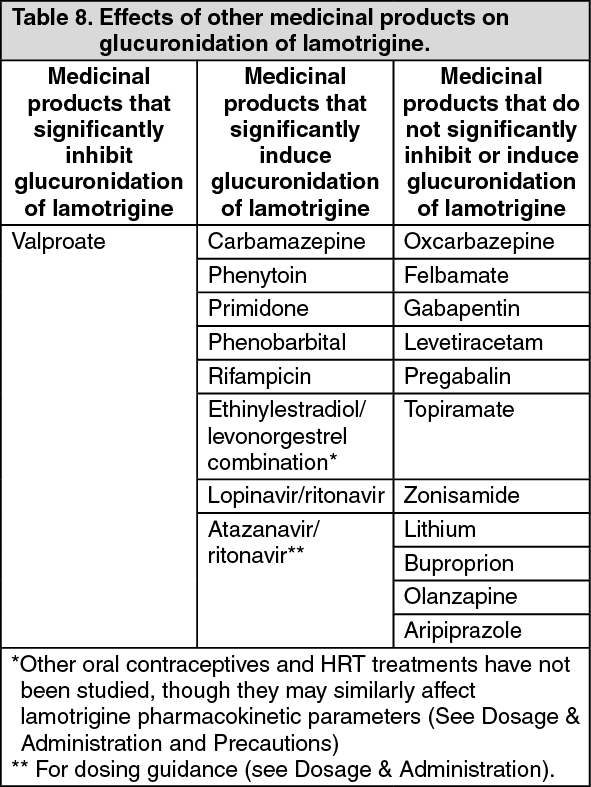 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image



 Sign Out
Sign Out