


 Sign Out
Sign Out
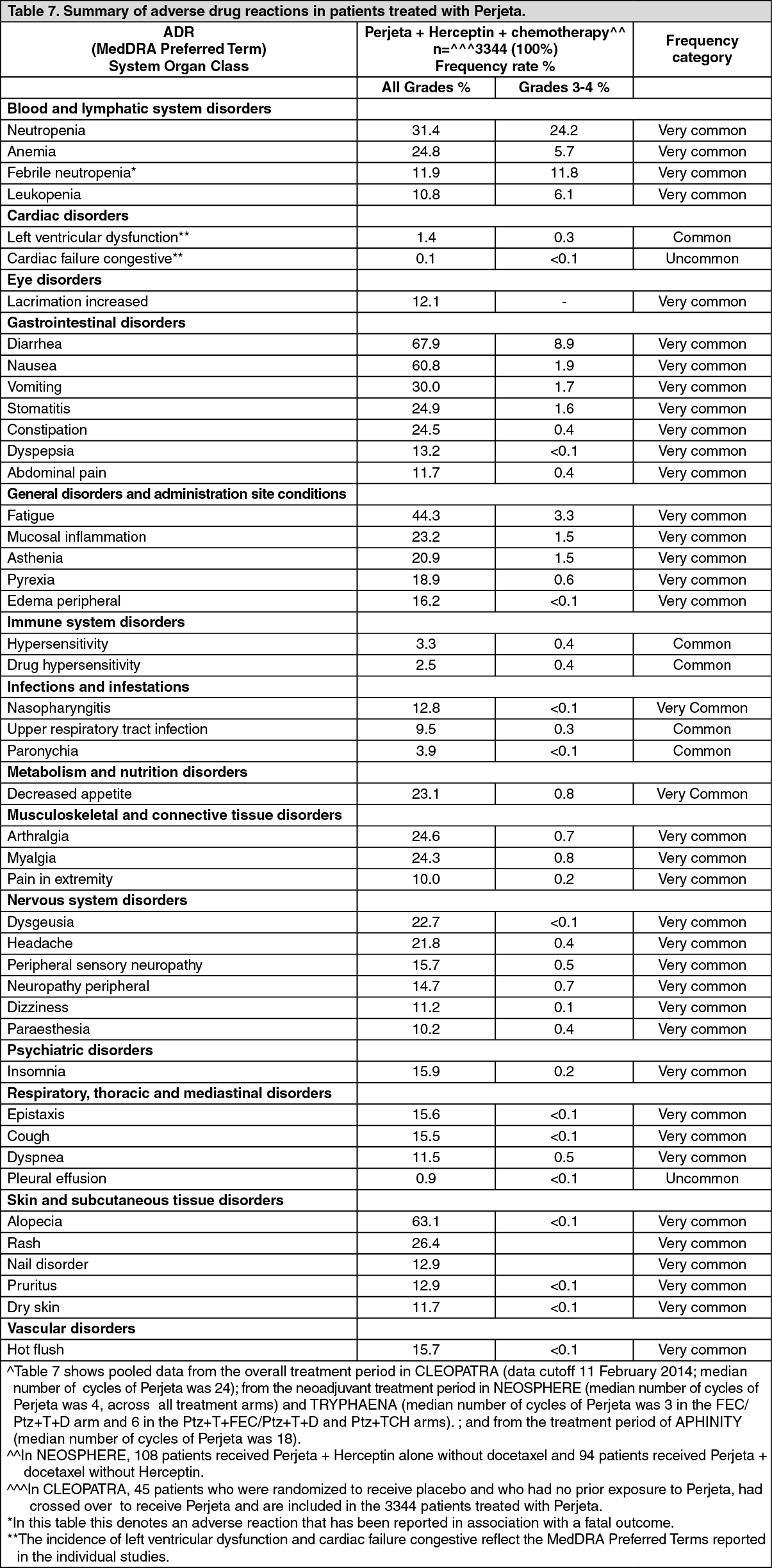
Metastatic and Early Breast Cancer: Table 7 summarizes the ADRs from the Perjeta-treatment arms of the following pivotal clinical trials: CLEOPATRA, in which Perjeta was given in combination with Herceptin and docetaxel to patients with MBC (n=453).
NEOSPHERE (n=309) and TRYPHAENA (n=218), in which neoadjuvant Perjeta was given in combination with Herceptin and chemotherapy to patients with locally advanced, inflammatory or EBC early breast cancer.
APHINITY, in which adjuvant Perjeta was given in combination with Herceptin and anthracycline-based or non-anthracycline-based, taxanecontaining chemotherapy to patients with EBC (n=2364).
As Perjeta is used with Herceptin and chemotherapy, it is difficult to ascertain the causal relationship of an adverse reaction to a particular drug.
The following categories of frequency have been used: very common (≥1/10), common (≥1/100 to <1/10), uncommon (≥1/1,000 to <1/100), rare (≥1/10,000 to <1/1000), very rare (<1/10,000).
The most common ADRs (≥30%) from this pooled data were diarrhea, alopecia, nausea, fatigue, neutropenia, and vomiting. The most common NCI-CTCAE Grade 3-4 ADRs (≥10%) were neutropenia and febrile neutropenia. (See Table 7.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageFurther information on selected adverse drug reactions: Left ventricular dysfunction: In the pivotal trial CLEOPATRA, the incidence of LVD during study treatment was higher in the placebo treated group than the Perjeta-treated group (8.6% and 6.6%, respectively). The incidence of symptomatic LVD was also lower in the Perjeta treated group (1.8% in the placebo treated group vs. 1.5% in the Perjeta treated group) (see Precautions).
In NEOSPHERE, in which patients received four cycles of Perjeta as neoadjuvant treatment, the incidence of LVD (during the overall treatment period) was higher in the Perjeta, Herceptin and docetaxel-treated groups (7.5%) compared to the Herceptin and docetaxel-treated group (1.9%). There was one case of symptomatic LVD in the Perjeta and Herceptin-treated group.
In TRYPHAENA the incidence of LVD (during the overall treatment period) was 8.3% in the group treated with Perjeta plus Herceptin and 5-fluorouracil, epirubicin and cyclophosphamide (FEC) followed by Perjeta plus Herceptin and docetaxel; 9.3% in the group treated with Perjeta plus Herceptin and docetaxel following FEC; and 6.6% in the group treated with Perjeta in combination with TCH. The incidence of symptomatic LVD (congestive heart failure) was 1.3% in the group treated with Perjeta plus Herceptin and docetaxel following FEC (this excludes a patient that experienced symptomatic LVD during FEC treatment prior to receiving Perjeta plus Herceptin and docetaxel) and also 1.3% in the group treated with Perjeta in combination with TCH. No patients in the group treated with Perjeta plus Herceptin and FEC followed by Perjeta plus Herceptin and docetaxel experienced symptomatic LVD.
In the neoadjuvant period of the BERENICE trial, the incidence of NYHA Class III/IV symptomatic LVD (congestive heart failure according to NCI-CTCAE v.4) was 1.5% in the group treated with dose dense AC followed by Perjeta plus Herceptin and paclitaxel and none of the patients (0%) experienced symptomatic LVD in the group treated with FEC followed by Perjeta in combination with Herceptin and docetaxel. The incidence of asymptomatic LVD (PT ejection fraction decrease according to NCI-CTCAE v.4) was 7% in the group treated with dose dense AC followed by Perjeta plus Herceptin and paclitaxel and 3.5% in the group treated with FEC followed by Perjeta plus Herceptin and docetaxel.
In APHINITY, the incidence of symptomatic heart failure (NYHA class III or IV) with a LVEF decline of at least 10% from baseline and to <50% was <1% (0.6% of Perjeta-treated patients vs 0.2% of placebo-treated patients). Of the patients who experienced symptomatic heart failure, 46.7% of Perjeta-treated patients and 66.8% of placebo-treated patients had recovered (defined as 2 consecutive LVEF measurements above 50%) at the data cutoff. The majority of the events were reported in anthracycline-treated patients. Asymptomatic or mildly symptomatic (NYHA class II) declines in LVEF of at least 10% from baseline and to <50% were reported in 2.7% of Perjeta-treated patients and 2.8% of placebo treated patients, of whom 79.7% of Perjeta-treated patients and 80.6% of placebo-treated patients had recovered at the data cutoff.
Infusion-related reactions: An infusion-related reaction was defined in the pivotal trials as any event reported as hypersensitivity, anaphylactic reaction, acute infusion reaction or cytokine release syndrome occurring during an infusion or on the same day as the infusion. In the pivotal trial CLEOPATRA, the initial dose of Perjeta was given the day before Herceptin (trastuzumab) and docetaxel to allow for the examination of Perjeta associated reactions. On the first day when only Perjeta was administered, the overall frequency of infusion related reactions was 9.8% in the placebo treated group and 13.2% in the Perjeta-treated group, with the majority of reactions being mild or moderate. The most common infusion related reactions (≥1.0%) in the Perjeta-treated group were pyrexia, chills, fatigue, headache, asthenia, hypersensitivity and vomiting.
During the second cycle when all drugs were administered on the same day, the most common infusion related reactions (≥1.0%) in the Perjeta-treated group were fatigue, drug hypersensitivity, dysgeusia, hypersensitivity, myalgia and vomiting (see Precautions).
In neoadjuvant and adjuvant trials, Perjeta was administered on the same day as the other study treatment drugs. Infusion-related reactions occurred in 18.6%-25.0% of patients on the first day of Perjeta administration (in combination with Herceptin and chemotherapy). The type and severity of events were consistent with those observed in CLEOPATRA, with a majority of reactions being mild or moderate.
Hypersensitivity reactions/anaphylaxis: In the pivotal trial CLEOPATRA the overall frequency of hypersensitivity/anaphylaxis reported events was 9.3% in the placebo-treated patients and 11.3% in the Perjeta-treated patients, of which 2.5% and 2.0% were NCI-CTCAE (version 3) grade 3-4, respectively. Overall, 2 patients in placebo-treated group and 4 patients in the Perjeta-treated group experienced anaphylaxis (see Precautions).
Overall, the majority of hypersensitivity reactions was mild or moderate in severity and resolved upon treatment. Based on modifications made to study treatment, most reactions were assessed as secondary to docetaxel infusions.
In neoadjuvant and adjuvant trials, hypersensitivity/anaphylaxis events were consistent with those observed in CLEOPATRA. In NEOSPHERE, two patients in the Perjeta and docetaxel treated group experienced anaphylaxis. In both TRYPHAENA and APHINITY, the overall frequency of hypersensitivity/anaphylaxis was highest in the Perjeta and TCH treated group (13.2% and 7.6% respectively), of which 2.6% and 1.3% of events, respectively were NCI-CTCAE Grade 3-4.
Laboratory Abnormalities: In the pivotal trials CLEOPATRA, NEOSPHERE, and APHINITY the incidence of NCI-CTCAE Grade 3-4 decreases in neutrophil counts were balanced in the Perjeta-treated and control groups.
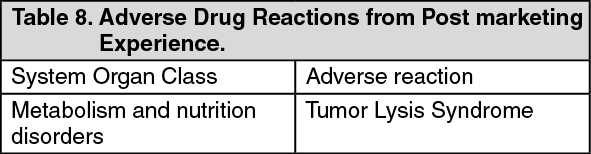
Postmarketing Experience: The following adverse drug reaction has been identified from post marketing experience with Perjeta based on spontaneous case reports and literature cases. The adverse drug reaction is listed according to system organ class in MedDRA. (See Table 8.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageLaboratory Abnormalities: Laboratory abnormalities reported from the post marketing setting are consistent with data from clinical trials of Perjeta.
View ADR Monitoring Form