Pharmacotherapeutic Group: Antineoplastic agent, recombinant humanized IgG1 monoclonal antibody; HER2 dimerization inhibitor.
ATC Code: L01XC13.
Pharmacology: Pharmacodynamics: Mechanism of Action: Perjeta is a recombinant humanized monoclonal antibody that specifically targets the extracellular dimerization domain (Subdomain II) of the human epidermal growth factor receptor 2 protein (HER2) and thereby blocks ligand-dependent heterodimerization of HER2 with other HER family members, including EGFR, HER3 and HER4. As a result, Perjeta inhibits ligand-initiated intracellular signaling through two major signal pathways, mitogen-activated protein (MAP) kinase and phosphoinositide 3-kinase (PI3K). Inhibition of these signaling pathways can result in cell growth arrest and apoptosis, respectively. In addition, Perjeta mediates antibody-dependent cell-mediated cytotoxicity (ADCC).
While Perjeta alone inhibited the proliferation of human tumor cells, the combination of Perjeta and Herceptin (trastuzumab) significantly augmented anti-tumor activity in HER2-overexpressing xenograft models.
Clinical/Efficacy Studies: HER2 overexpression was determined at a central laboratory and defined as a score of 3+ by IHC or an ISH amplification ratio ≥2.0 in the trials as follows.
Metastatic Breast Cancer: Perjeta in Combination with Herceptin (trastuzumab) and docetaxel: CLEOPATRA is a multicenter, randomized, double-blind, placebo-controlled Phase III clinical trial conducted in 808 patients with HER2-positive metastatic or locally recurrent unresectable breast cancer who have not received previous anti-HER2 therapy or chemotherapy for their metastatic disease. Patients were randomized 1:1 to receive placebo plus Herceptin (trastuzumab) and docetaxel or Perjeta plus Herceptin (trastuzumab) and docetaxel. Randomization was stratified by prior treatment status (de novo or prior adjuvant/neoadjuvant therapy) and geographic region (Europe, North America, South America and Asia). Patients with prior adjuvant or neoadjuvant therapy were required to have a disease free interval of at least 12 months before enrolment into the trial.
Perjeta and Herceptin were administered intravenously as outlined in Dosage & Administration. Patients were treated with Perjeta and Herceptin (trastuzumab) until disease progression, withdrawal of consent or unmanageable toxicity. Docetaxel was given as an initial dose of 75 mg/m
2 IV infusion every 3 weeks for at least 6 cycles. The dose of docetaxel could be escalated to 100 mg/m
2 at the investigator's discretion if the initial dose was well tolerated.
At the time of the primary analysis, the mean number of cycles of study treatment received was 16.2 in the placebo treatment group and 19.9 in the Perjeta treated group.
The primary endpoint of the study was progression-free survival (PFS) as assessed by an independent review facility (IRF) and defined as the time from the date of randomization to the date of disease progression or death (from any cause) if the death occurred within 18 weeks of the last tumor assessment. Secondary efficacy endpoints were overall survival (OS), PFS (investigator-assessed), objective response rate (ORR), duration of response, and time to symptom progression according to the FACT B QoL questionnaire.
Demographics were well balanced (median age was 54 years old, the majority were Caucasian (59%) and all were female with the exception of 2 patients). Approximately half the patients in each treatment group had hormone receptor-positive disease (defined as estrogen receptor [ER] positive and/or progesterone receptor [PgR] positive) and approximately half of the patients in each treatment group had received prior adjuvant or neo-adjuvant therapy (192 patients [47.3%] in the placebo treated group vs 184 patients [45.8%] Perjeta treated group).
At the time of the primary progression-free survival analysis, a total of 242 patients (59%) in the placebo treated group and 191 patients (47.5%) in the Perjeta treated group had IRF-confirmed progressive disease or had died within 18 weeks of their last tumor assessment.
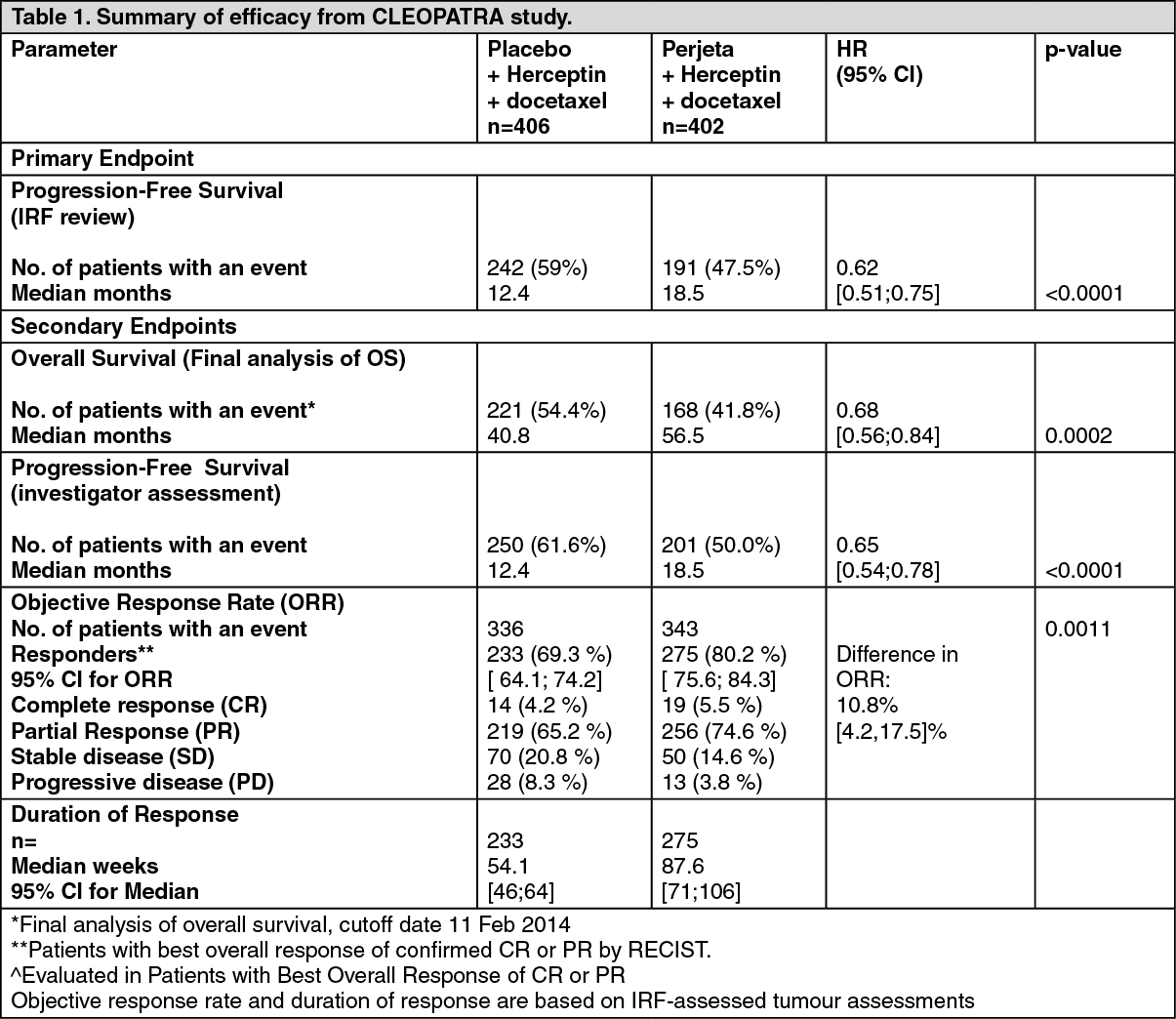
At the time of the primary analysis the CLEOPATRA study demonstrated a statistically significant improvement in IRF-assessed PFS (hazard ratio [HR] = 0.62, 95% CI = 0.51, 0.75, p<0.0001) in the Perjeta treated group compared with the placebo treated group, and an increase in median PFS of 6.1 months (median PFS of 12.4 months in the placebo treated group vs 18.5 months in the Perjeta treated group) (see Figure 1). The results for investigator-assessed PFS were comparable to those observed for IRF-assessed PFS (median PFS was 12.4 months for placebo vs 18.5 months for Perjeta) (see Table 1). Consistent results were observed across pre-specified patient subgroups including the subgroups based on stratification factors of geographic region and prior adjuvant/neoadjuvant therapy or de novo metastatic breast cancer (see Figure 2).
The efficacy results from the CLEOPATRA trial are summarized in Table 1. (See Table 1 and Figure 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
At the primary analysis of efficacy an interim analysis of OS showed a strong trend suggestive of a survival benefit in favor of the Perjeta treated group.
An interim analysis of OS performed one year after the primary analysis of efficacy, demonstrated a statistically significant OS benefit in favor of the Perjeta-treated group (HR 0.66, p = 0.0008 log-rank test). The median time to death was 37.6 months in the placebo-treated group but had not yet been reached in the Perjeta-treated group.
The final analysis of OS was performed when 389 patients had died (221 in the Placebo-treated group and 168 in the Perjeta-treated group). The statistically significant OS benefit in favor of the Perjeta-treated group was maintained (HR 0.68, p = 0.0002 log-rank test). The median time to death was 40.8 months in the placebo-treated group and 56.5 months in the Perjeta-treated group (see Table 1, Figures 2 and 3).
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
There was no statistically significant difference between treatment groups in Health Related Quality of Life as assessed by time to symptom progression on the FACT-B TOI-PFB subscale, defined as a 5 point reduction in subscale score (HR =0.97, 95% CI =0.81; 1.16). In an exploratory analysis, patients treated with Perjeta in combination with Herceptin (trastuzumab) and docetaxel experienced a lower risk of symptom progression on the FACT-B breast cancer subscale (defined as a 2 point reduction in subscale score) compared to those treated with Herceptin (trastuzumab) and docetaxel alone (HR =0.78, 95% CI =0.65; 0.94).
BO17929: BO17929 was a Phase II, single arm, non-randomized study with Perjeta and was conducted in patients with HER2-positive MBC who had received prior treatment with Herceptin (trastuzumab).
The trial was divided into 3 cohorts.
Cohorts 1 and 2: 66 patients in cohorts 1 and 2 received at least one dose of Perjeta and Herceptin (trastuzumab) (all treated population and all patients had received prior treatment for metastatic disease; half were receiving second-line treatment for metastatic disease, while 35% were receiving third-line treatment and beyond. In addition, 71% had received neoadjuvant chemotherapy). At the time of the primary analysis, the median duration of treatment on study was nine cycles (27 weeks). At the time of the primary analysis, the ORR and CBR are presented in Table 2. The median PFS and time to progression (TTP) were 24 weeks. Median time to response was 11 weeks, and in those patients with a response, the median duration of response was 25 weeks.
Cohort 3: 29 patients received at least one cycle of Perjeta, of these 29 patients, 12 participated in the single-agent Phase only, and 17 went on to receive Perjeta and Herceptin (trastuzumab) treatment when they had documented progression on Perjeta alone. All 29 patients had progressed on first-line therapy in the metastatic setting, and 41.4% had also progressed after second line therapy. All patients in Cohort 3 received at least one full dose of Perjeta. Patients on Perjeta and Herceptin (trastuzumab) treatment received a median of 12 cycles overall. Table 2 shows that Perjeta alone had modest activity in patients after failure of Herceptin (trastuzumab) (middle column). These responses occurred in patients whose disease had recently progressed on each antibody when given separately. In addition 3 patients had stable disease lasting six months or longer for a total clinical benefit rate of 35.3%. (See Table 2.)
Click on icon to see table/diagram/image
Early Breast Cancer: NEOSPHERE (WO20697): NEOSPHERE is a multicenter, randomized Phase II clinical trial conducted in 417 patients with operable, locally advanced, or inflammatory HER2-positive breast cancer (T2-4d) who were scheduled for neoadjuvant therapy. Breast Patients were randomized to receive one of four neoadjuvant regimens prior to surgery as follows: Herceptin plus docetaxel, Perjeta plus Herceptin and docetaxel, Perjeta plus Herceptin, or Perjeta plus docetaxel. Randomization was stratified by breast cancer type (operable, locally advanced, or inflammatory) and estrogen (ER) or progesterone (PgR) positivity.
Perjeta and Herceptin were administered intravenously (see Dosage & Administration) for 4 cycles. Following surgery all patients received three cycles of 5-Fluorouracil (600 mg/m
2), epirubicin (90 mg/m
2), cyclophosphamide (600 mg/m
2) (FEC) given intravenously every three weeks and Herceptin administered intravenously every three weeks to complete one year of therapy Patients in the Perjeta plus Herceptin and docetaxel arm received docetaxel every three weeks for four cycles prior to FEC after surgery so that all patients received equivalent cumulative doses of the chemotherapeutic agents and Herceptin.
The primary endpoint of the study was pathological complete response (pCR) rate in the breast (ypT0/is). Secondary efficacy endpoints were clinical response rate, breast conserving surgery rate (T2-3 only), disease-free survival (DFS), and PFS [46]. Additional exploratory pCR rates included nodal status (ypT0/isN0 and ypT0N0).
Demographics were well balanced (median age was 49-50 years old, the majority were Caucasian (71%)) and all were female. Overall 7% of patients had inflammatory breast cancer, 32% had locally advanced breast cancer and 61% had operable breast cancer. Approximately half the patients in each treatment group had hormone receptor-positive disease (defined as ER positive and/or PgR positive).
The efficacy results are summarized in Table 3. A statistically significant and clinically meaningful improvement in pCR rate (ypT0/is) was observed in patients receiving Perjeta plus Herceptin and docetaxel compared to patients receiving Herceptin and docetaxel (45.8% vs 29.0%, p value = 0.0141). A consistent pattern of results was observed regardless of pCR definition.
Pathological complete response (pCR) rates as well as the magnitude of improvement with Perjeta were lower in the subgroup of patients with hormone receptor-positive tumors than in patients with hormone receptor-negative tumors (5.9% to 26.0% and 27.3% to 63.2%, respectively).
TRYPHAENA (BO22280): TRYPHAENA is a multicenter, randomized Phase ll clinical study conducted in 225 patients with HER2-positive locally advanced, operable, or inflammatory (T2-4d) breast cancer. Patients were randomized to receive one of three neoadjuvant regimens prior to surgery as follows: 3 cycles of FEC followed by 3 cycles of docetaxel all in combination with Perjeta and Herceptin, 3 cycles of FEC alone followed by 3 cycles of docetaxel and Herceptin in combination with Perjeta, or 6 cycles of TCH in combination with Perjeta. Randomization was stratified by breast cancer type (operable, locally advanced, or inflammatory) and ER and/or PgR positivity.
Perjeta and Herceptin were administered intravenously as outlined in Dosage & Administration. 5-Fluorouracil (500 mg/m
2), epirubicin (100 mg/m
2), cyclophosphamide (600 mg/m
2) were given intravenously every three weeks for 3 cycles. Docetaxel was given as an initial dose of 75 mg/m
2 IV infusion every three weeks with the option to escalate to 100 mg/m
2 at the investigator's discretion if the initial dose was well tolerated. However, in the Perjeta in combination with TCH arm, docetaxel was given intravenously at 75 mg/m
2 and no escalation was permitted and carboplatin (AUC 6) was given intravenously every three weeks. Following surgery all patients received Herceptin to complete one year of therapy, which was administered intravenously every 3 weeks.
The primary endpoint of this study was cardiac safety during the neoadjuvant treatment period of the study. Secondary efficacy endpoints were pCR rate in the breast (ypT0/is), DFS, PFS and OS.
Demographics were well balanced (median age was 49-50 years old, the majority were Caucasian (77%)) and all were female. Overall 6% of patients had inflammatory breast cancer, 25% had locally advanced breast cancer and 69% had operable breast cancer, with approximately half the patients in each treatment group had ER-positive and/or PgR-positive disease.
High pCR rates were observed in all 3 treatment arms (see Table 3). A consistent pattern of results was observed regardless of pCR definition. pCR rates were lower in the subgroup of patients with hormone receptor-positive tumors than in patients with hormone receptor-negative tumors (46.2% to 50.0% and 65.0% to 83.8% respectively). (See Table 3.)
Click on icon to see table/diagram/image
BERENICE (WO29217): BERENICE is a non-randomized, open-label, multicenter, multinational, Phase II trial conducted in 401 patients with HER2-positive locally advanced, inflammatory, or early-stage HER2-positive breast cancer.
The BERENICE study included two parallel groups of patients. Patients considered suitable for neoadjuvant treatment with Herceptin plus anthracycline/taxane-based chemotherapy were allocated to receive one of the two following regimens prior to surgery as follows: Cohort A - 4 cycles of two weekly doxorubicin and cyclophosphamide (dose dense AC) followed by 4 cycles of Perjeta in combination with Herceptin and paclitaxel.
Cohort B - 4 cycles of FEC followed by 4 cycles of Perjeta in combination with Herceptin and docetaxel.
Perjeta and Herceptin were administered intravenously as outlined in Dosage & Administration. Doxorubicin 60 mg/m
2 IV and cyclophosphamide 600 mg/m
2 IV were administered every 2 weeks (ddAC) for four cycles (Cycles 1-4) with G-CSF (granulocyte colony stimulating factor) support at investigator discretion, followed by paclitaxel 80 mg/m
2 IV weekly for 12 weeks (Cycles 5-8), with Perjeta and Herceptin every 3 weeks during Cycles 5-8 (from the start of paclitaxel; four cycles of Perjeta and Herceptin in total during the neoadjuvant period). 5-Fluorouracil (500 mg/m
2), epirubicin (100 mg/m
2), cyclophosphamide (600 mg/m
2) were given intravenously every three weeks for 4 cycles. Docetaxel was given at an initial dose of 75 mg/m
2 IV infusion every three weeks with the option to escalate to 100 mg/m
2 at the investigator's discretion if the initial dose was well tolerated. Following surgery all patients received Perjeta and Herceptin which were administered intravenously every 3 weeks, to complete one year of therapy.
The primary endpoint of this study was cardiac safety during the neoadjuvant treatment period of the study (see Adverse Reactions). Key secondary endpoints at the time of primary analysis were neoadjuvant safety and pCR rate in the breast and nodes (i.e. ypT0/is ypN0). Long-term clinical and safety outcomes will also be assessed (IDFS, EFS and OS, not yet available).
Demographics of the patients were well balanced between the groups. The median age of the patients was 49 years, the majority of patients were Caucasian (83%) and all but one patient was female. Approximately two-thirds of patients (64.3% [n = 128] in Cohort A and 61.7% [n = 124] in Cohort B) had hormone receptor-positive disease.
High pCR rates were observed in both treatment arms, with pCR (ypT0/is ypN0) rates of 61.8% in Cohort A and 60.7% in Cohort B. A consistent pattern of results was observed regardless of pCR definition. pCR rates were lower in the subgroup of patients with hormone receptor-positive tumors than in patients with hormone receptor-negative tumors in both Cohorts (51.6% to 81.5% and 57.3% to 68.0% respectively).
APHINITY (BO25126): APHINITY is a multicenter, randomized, double-blind, placebo-controlled Phase III trial conducted in 4804 patients with HER2-positive early breast cancer who had their primary tumor excised prior to randomization. Patients were then randomized to receive Perjeta or placebo, in combination with adjuvant Herceptin and chemotherapy. Investigators selected one of the following anthracycline-based or non-anthracycline-based chemotherapy regimens for individual patients: 3 or 4 cycles of FEC or 5-fluorouracil, doxorubicin and cyclophosphamide (FAC), followed by 3 or 4 cycles of docetaxel or 12 cycles of weekly paclitaxel.
4 cycles of AC or EC, followed by 3 or 4 cycles of docetaxel or 12 cycles of weekly paclitaxel.
6 cycles of docetaxel in combination with carboplatin.
Perjeta and Herceptin were administered intravenously (see Dosage & Administration) every 3 weeks starting on Day 1 of the first taxane-containing cycle, for a total of 52 weeks (maximum 18 cycles) or until recurrence, withdrawal of consent or unmanageable toxicity. Standard doses of 5-fluorouracil, epirubicin, doxorubicin, cyclophosphamide, docetaxel, paclitaxel and carboplatin were administered. After completion of chemotherapy, patients received radiotherapy and/or hormone therapy as per local clinical standard.
The primary endpoint of the study was invasive disease-free survival (IDFS), defined as the time from randomization to first occurrence of ipsilateral local or regional invasive breast cancer recurrence, distant recurrence, contralateral invasive breast cancer, or death from any cause.
Demographics were well balanced between the two treatment arms. The median age was 51 years, and over 99% of patients were female. The majority of patients had node-positive (63%) and/or hormone receptor-positive disease (64%), and were Caucasian (71%).
After a median follow-up to 45.4 months, the APHINITY study demonstrated 19% (hazard ratio [HR] = 0.81) reduction in risk of recurrence or death in patients randomized to receive Perjeta compared with patients randomized to receive placebo.
The efficacy results from the APHINITY trial are summarized in Table 4 and in Figures 4 and 5. (See Table 4 and Figures 4 and 5.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The estimate of IDFS at 4-years was 92.3% in the Perjeta-treated group versus 90.6% in the placebo-treated group. At the time of the estimate the median follow-up was 45.4 months.
Results of Subgroup Analysis: Consistent results were observed across the majority of pre-specified patient subgroups. The benefits of Perjeta were more apparent for patients in certain high risk groups, notably patients with node-positive or hormone receptor negative disease.
Estimates of IDFS rates in the node positive subgroup were 92.0% versus 90.2% at 3 years and 89.9% vs. 86.7% at 4 years in Perjeta-treated patients versus the placebo-treated patients, respectively. In the node negative subgroup estimates of IDFS rates were 97.5% versus 98.4% at 3 years and 96.2% versus 96.7% at 4 years in Perjeta-treated patients versus placebo-treated patients, respectively. In the hormone receptor-positive subgroup estimates of IDFS were 94.8% versus 94.4% at 3 years and 93.0% versus 91.6% at 4 years in Perjeta-treated patients versus placebo-treated patients, respectively. In the hormone receptor-negative subgroup estimates of IDFS rates were 92.8% versus 91.2% at 3 years and 91.0% versus 88.7% at 4 years in Perjeta-treated patients versus placebo-treated patients, respectively.
Patient Reported Outcomes (PRO): Secondary endpoints included the assessment of patient-reported global health status, role and physical function, and treatment symptoms using the EORTC QLQ-C30 and EORTC QLQ-BR23 questionnaires. In the analyses of patient reported outcomes, a 10-point difference was considered clinically meaningful.
Patients' physical function, global health status and diarrhea scores showed a clinically meaningful change during chemotherapy in both treatment arms. The mean decrease from baseline at that time for physical function was -10.7 (95% CI -11.4, -10.0) in the Perjeta arm and -10.6 (95% -11.4, -9.9) in the placebo arm; global health status was -11.2 (95% CI -12.2, -10.2) in the Perjeta arm and -10.2 (95% CI -11.1,-9.2) in the placebo arm. Change in diarrhea symptoms increased to +22.3 (95% CI 21.0, 23.6) in the Perjeta arm versus +9.2 (95% CI 8.2, 10.2) in the placebo arm.
Thereafter in both arms, physical function and global health status scores returned to baseline levels during targeted treatment. Diarrhea symptoms returned to baseline after HER2 therapy in the Perjeta-arm. The addition of Perjeta to Herceptin plus chemotherapy did not affect patients' overall role function over the course of the study.
Click on icon to see table/diagram/image
Immunogenicity: Patients in the pivotal trial CLEOPATRA were tested at multiple time-points for anti-drug antibodies (ADA) to Perjeta. 6.7% (25/372) of patients in the placebo treated group and 3.3% (13/389) of patients in the Perjeta treated group tested positive for ADA. In BERENICE, 4.1% (16/392) of the patients treated with Perjeta tested positive for ADA. None of these patients experienced anaphylactic/hypersensitivity reactions that were clearly related to ADA.
Immunogenicity assay results are highly dependent on several factors including assay sensitivity and specificity, assay methodology, sample handling, timing of sample collection, concomitant medications and underlying disease. For these reasons, comparison of incidence of antibodies to Perjeta with the incidence of antibodies to other products may be misleading.
Pharmacokinetics: Across multiple clinical trials in various indications there was no change in clearance of pertuzumab at doses of 2-25 mg/kg. Based on a population PK analysis that included 481 patients, the median clearance (CL) of pertuzumab was 0.235 L/day and the median half-life was 18 days.
The population PK analysis suggested no PK differences based on age, gender and ethnicity (Japanese versus non-Japanese). Baseline albumin and lean body weight were the most significant covariates influencing CL. Clearance decreased in patients with higher baseline albumin concentrations and increased in patients with greater lean body weight. However sensitivity analyses performed at the recommended dose and schedule of Perjeta showed that at the extreme values of these two covariates, there was no significant impact on the ability to achieve target steady-state concentrations identified in preclinical tumor xenograft models. Therefore, there is no need to adjust the dosage of pertuzumab based on these covariates.
The PK results of pertuzumab in the NEOSPHERE study were consistent with the predictions from the previous population PK model. No differences in pertuzumab PK were observed in patients with early breast cancer compared to patients with metastatic breast cancer.
Absorption: Pertuzumab is administered as an IV infusion.
Distribution: Across all clinical studies, the volume of distribution of the central (Vc) and the peripheral (Vp) compartment in the typical patient, was 3.11 L and 2.46 L, respectively.
Metabolism: The metabolism of pertuzumab has not been directly studied. Antibodies are cleared principally by catabolism.
Elimination: The median clearance (CL) of pertuzumab was 0.235 L/day and the median half-life was 18 days.
Pharmacokinetics in Special Populations: Geriatric population: No dedicated studies of pertuzumab have been conducted in geriatric patients. In a population PK analysis, age was not found to significantly affect PK of pertuzumab. In the population PK analysis, 32.5% (N=143) patients were ≥65 years of age and 9.1% (N=40) patients were ≥75 years of age.
Renal impairment: No formal PK study has been conducted in patients with renal impairment. Based on the population PK analysis, renal impairment is not expected to influence pertuzumab exposure; however, only limited data from patients with moderate and severe renal impairment were included in the population.
Toxicology: Non-Clinical Safety: Carcinogenicity: Long-term studies in animals have not been performed to evaluate the carcinogenic potential of pertuzumab.
Genotoxicity: Studies have not been performed to evaluate the mutagenic potential of pertuzumab.
Impairment of Fertility: No specific fertility studies in animals have been performed to evaluate the effect of pertuzumab. No adverse effects on male and female reproductive organs were observed in repeat-dose toxicity studies of up to six month duration in cynomolgus monkeys.
Reproductive Toxicity: Reproductive toxicology studies have been conducted in cynomolgus monkeys at loading doses of 30 to 150 mg/kg and maintenance doses of 10 to 100 mg/kg achieving clinically relevant exposures. Intravenous administration of pertuzumab from Gestation Day (GD) 19 through 50 (period of organogenesis) has been shown to be embryotoxic with a dose dependent increase in embryo-fetal deaths between GD 25 to 70. Delayed renal development and oligohydramnios were identified at GD100.
Other: In cynomolgus monkeys, weekly IV administration of pertuzumab at doses up to 150 mg/kg/dose were generally well tolerated. With doses of 15 mg/kg and higher intermittent mild treatment-associated diarrhoea was noted. In a subset of monkeys chronic dosing (7 to 26 weekly doses) resulted in episodes of diarrhoea-related dehydration which were managed with intravenous fluid replacement therapy.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image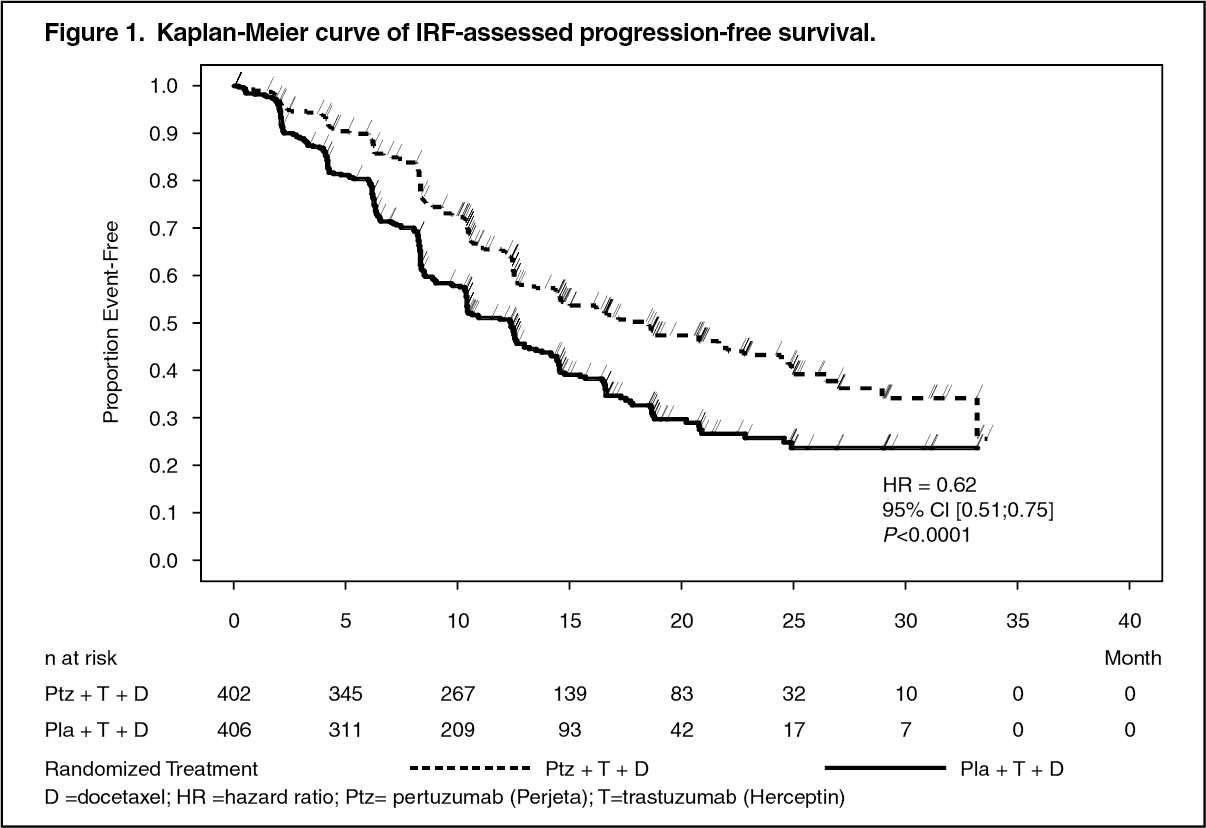 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image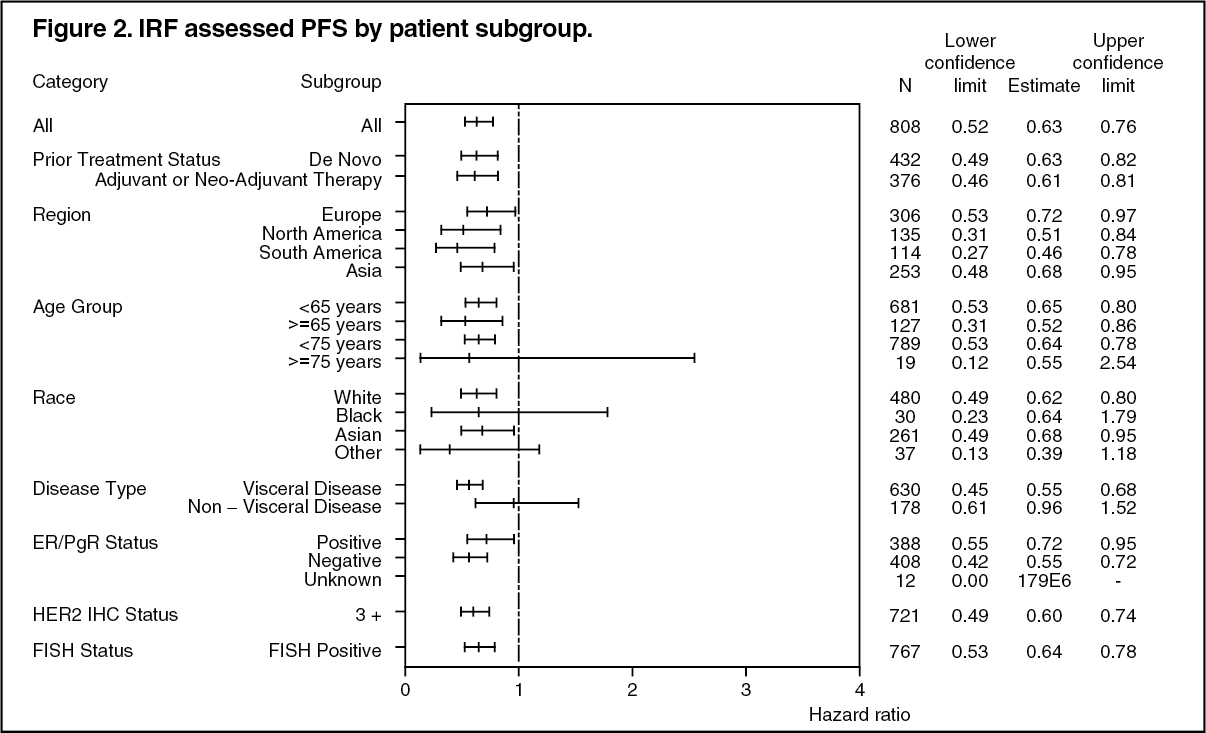 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image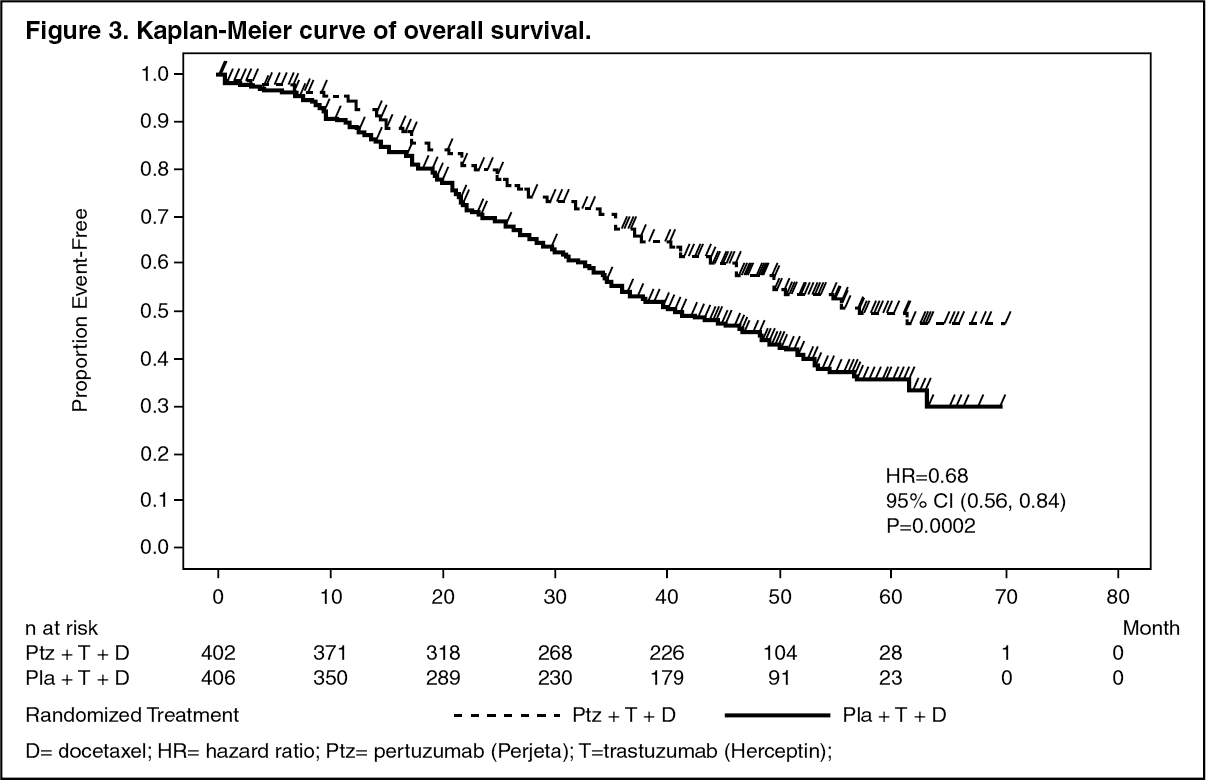 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image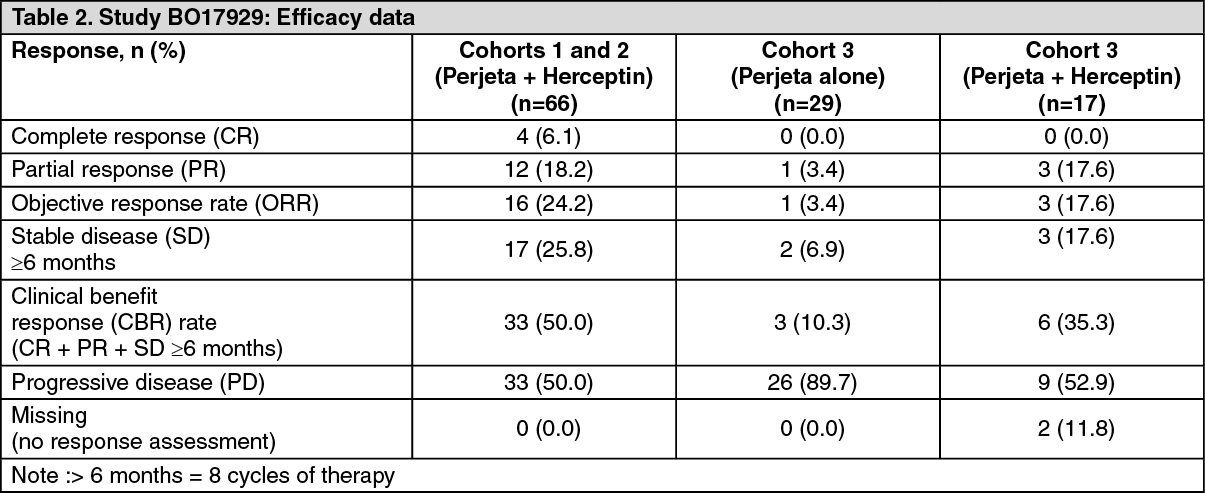 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image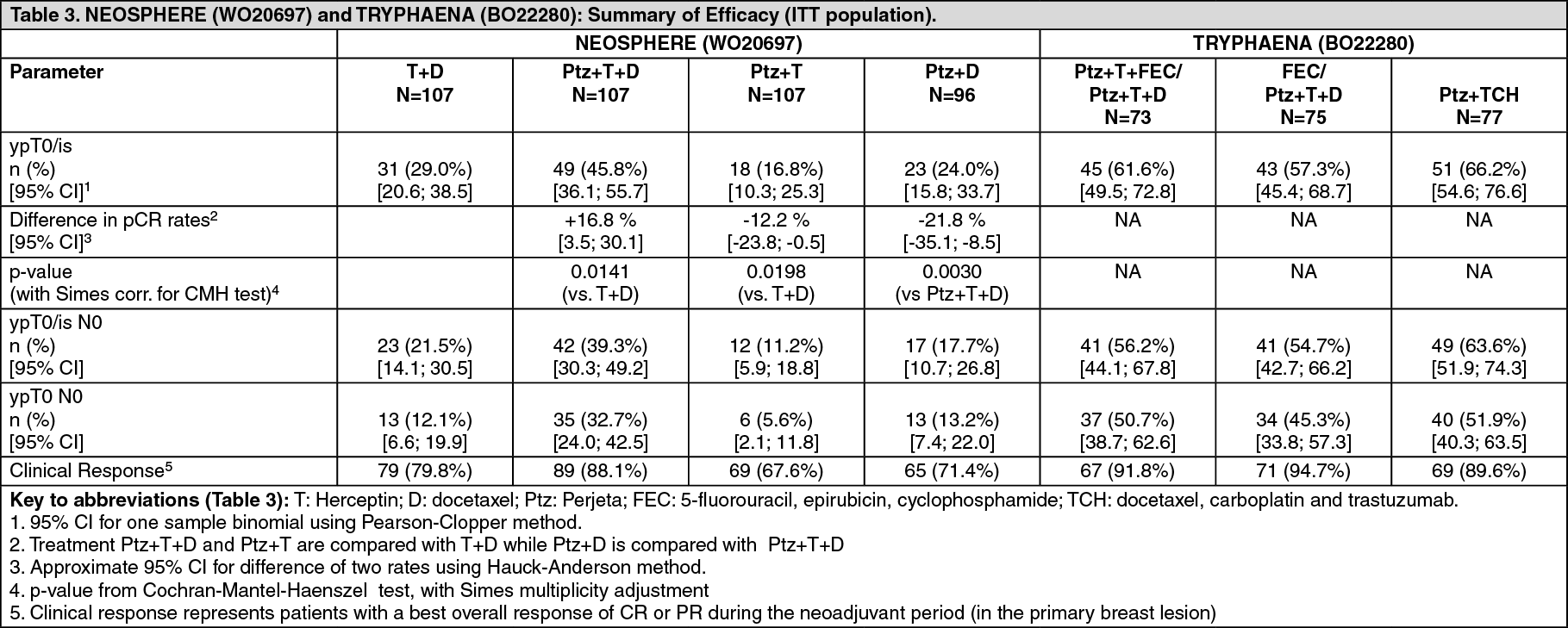 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image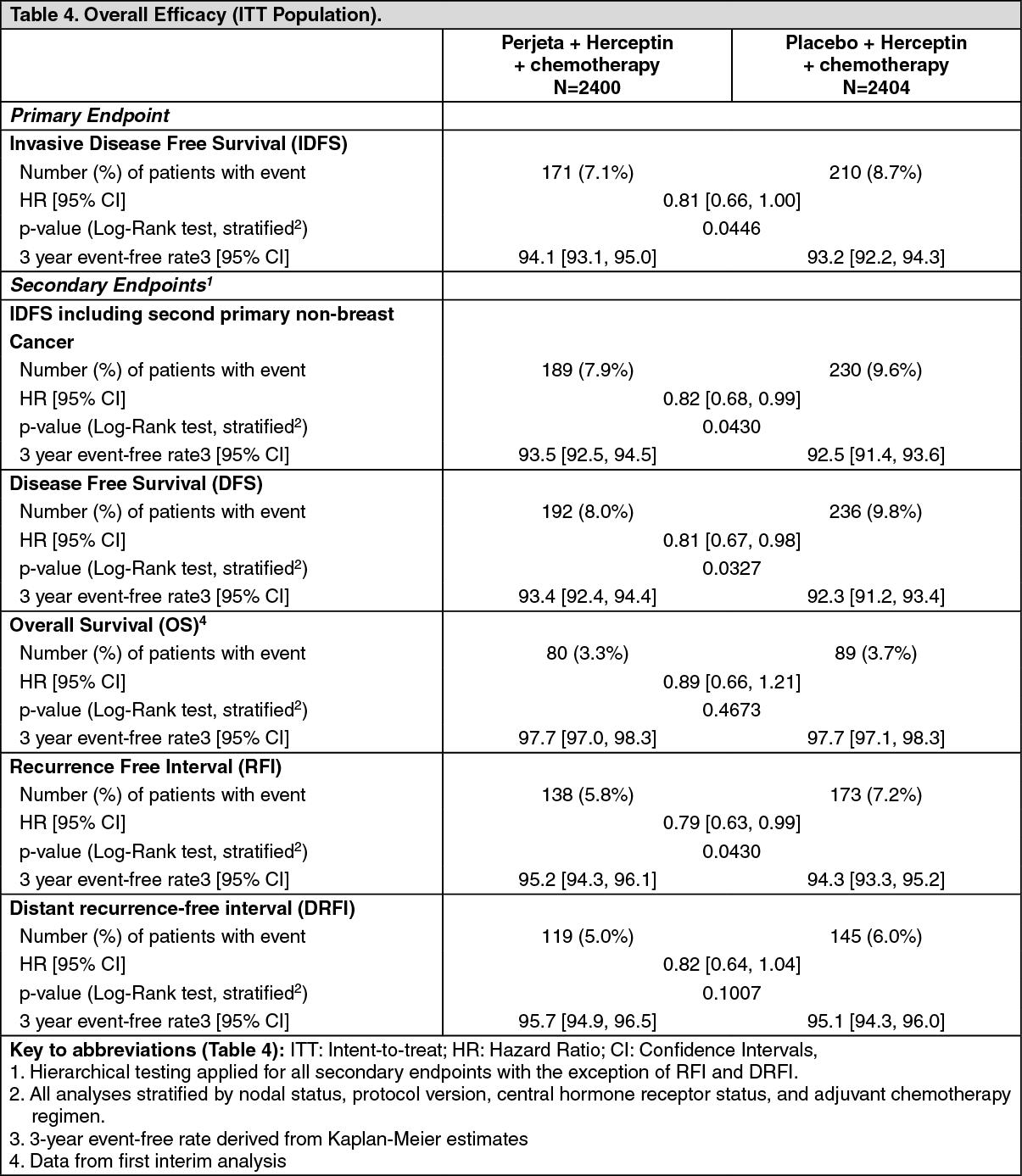 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image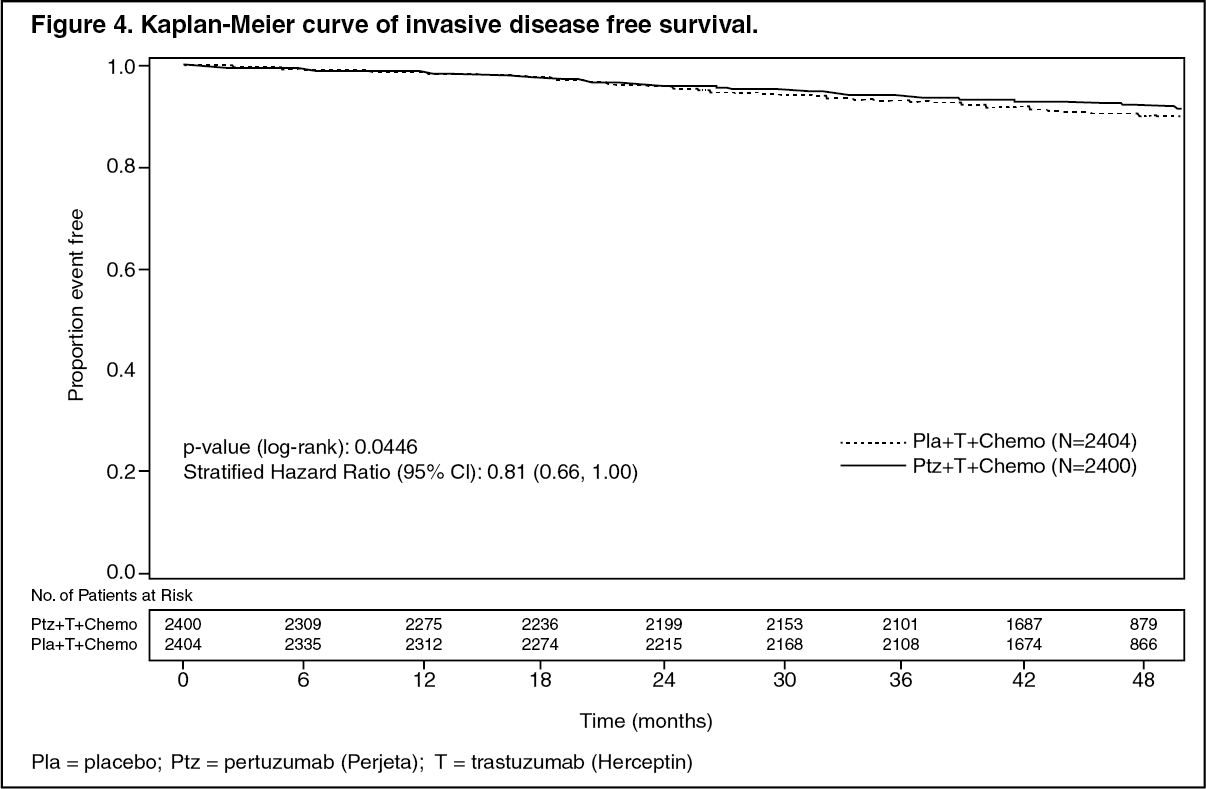 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image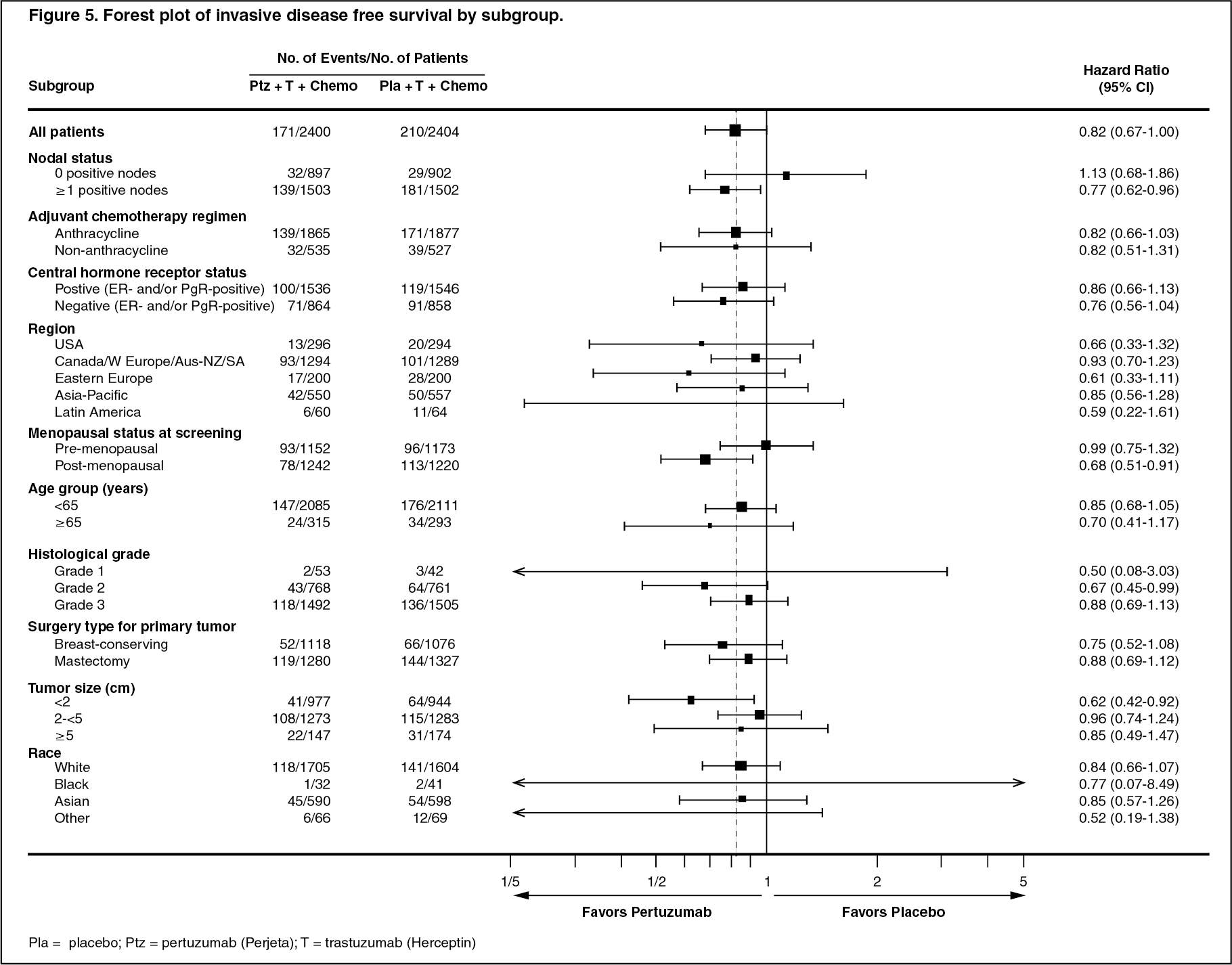 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image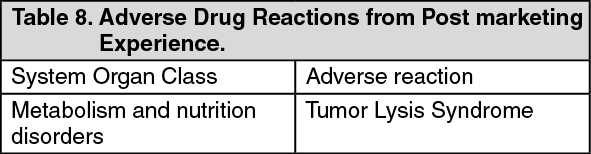 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out