


 Sign Out
Sign Out

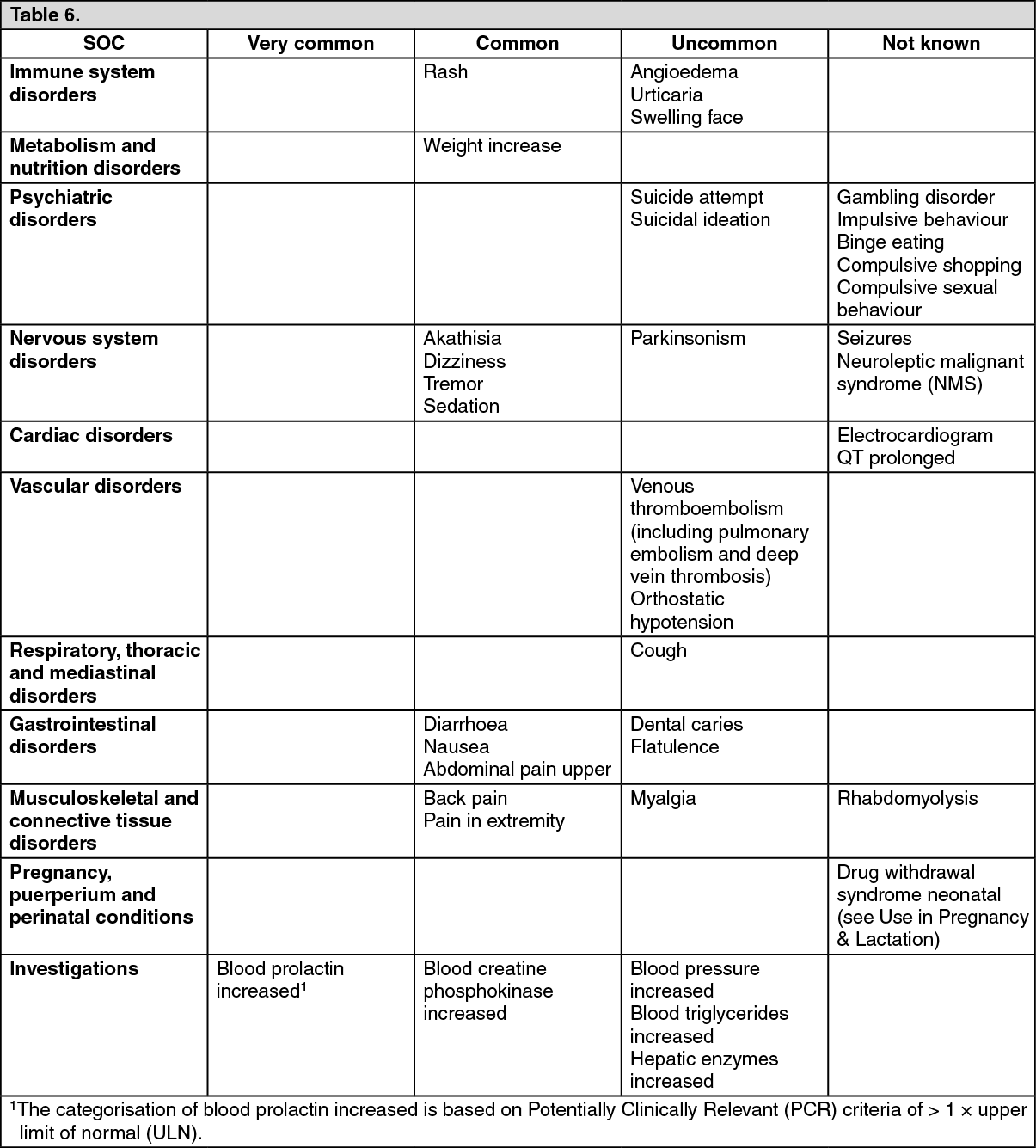
Tabulated list of adverse reactions: The incidences of the ADRs associated with brexpiprazole therapy are tabulated below. The table is based on adverse reactions reported in short-term placebo-controlled phase 2 and 3 clinical trials with relevant therapeutic doses (2 mg to 4 mg).
All ADRs are listed by system organ class (SOC) and frequency: very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 to < 1/1,000), very rare (< 1/10,000) and not known (cannot be estimated from the available data). Within each frequency grouping, adverse reactions are presented in order of decreasing seriousness. (See Table 6.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageDescription of selected adverse reactions: Extrapyramidal Symptoms (EPS): Akathisia was the most frequently reported EPS related ADR in the brexpiprazole 2 mg/day to 4 mg/day group (5.6 %) compared to 4.5 % in placebo, followed by tremor (2.7 %) compared to 1.2 % in placebo. The incidences of other EPS-related ADRs reported in short-term, controlled trials are dyskinesia (0.4 %), extrapyramidal disorder (1.8 %) and Parkinsonism (0.4 %).
Akathisia: From fixed-dose trials there appears to be a dose-response relationship for akathisia in patients treated with brexpiprazole, with an increasing frequency with higher doses. The incidence of akathisia in the brexpiprazole 1 mg/day, 2 mg/day, and 4 mg/day groups was 3.0 %, 4.6 %, and 6.5 %, respectively, compared with 5.2 % of subjects in the placebo group.
The incidence of akathisia in the short-term, controlled trials (5.4 %) was similar to the incidence in the long-term, open-label trials (5.7 %).
Suicidality: In short-term, controlled trials, Treatment Emergent Adverse Events (TEAEs) related to suicidality were reported for 8 subjects (0.5 %, 2 serious events, 1 leading to discontinuation) in the all brexpiprazole treatment group and 3 subjects (0.4 %, none serious) in the placebo group. In long-term, open-label trials, TEAEs related to suicidality were reported for 23 subjects (1.6 %). Overall in the brexpiprazole clinical development program for schizophrenia, one death due to suicide, considered not drug related by the investigator, occurred. Spontaneous cases reporting completed suicide and suicide attempt have been reported in the post-marketing setting.
QT prolongation: In the short-term controlled trials with brexpiprazole, 3 TEAEs related to QT prolongation were reported in the 2 mg to 4 mg group (0.3 %), compared with 3 TEAEs (0.5 %) reported for subjects on placebo. The incidence of TEAEs in long-term trials was similar to that of the short-term trials. The effects of brexpiprazole at therapeutic (4 mg) and supra-therapeutic (12 mg) doses on QT interval were evaluated in subjects with schizophrenia or schizoaffective disorder in a randomised, double-blind, placebo- and positive-controlled (moxifloxacin), parallel-arm trial. Subgroup analyses from this trial suggested that the QTc prolongation was larger in female subjects than in males (see Pharmacology: Pharmacodynamics under Actions).
Weight gain: In short-term, controlled trials, the percentage of subjects with clinically significant weight gain (increase of ≥ 7 % from baseline in body weight) was 9.1 % in the brexpiprazole 2 mg/day to 4 mg/day group, compared with 3.8 % in the placebo group.
In the long-term, open-label trial, the percentage of subjects with clinically significant weight gain (increase of ≥ 7 % in body weight) at any visit was 20.7 % and 0.4 % of the subjects discontinued due to weight gain. In subjects who had a weight gain ≥ 7 % from baseline, weight increased over time, with mean weight gain up to 10.2 kg at week 52. The mean change in body weight overall for the brexpiprazole group in the long term, open label trial was 2.1 kg at week 52.
Prolactin: The incidence of blood prolactin increased was 0.9 % in 2 mg to 4 mg brexpiprazole group compared to that of 0.5 % in placebo in short-term, controlled trials. Higher frequencies of prolactin increased (1.5 % versus 0.60 %) were observed in females compared to males in short-term trials. In addition, the frequencies of prolactin elevations > 1 × ULN in the 2 mg to 4 mg brexpiprazole group was 13.7 % in females versus 6.4 % in placebo and 11.1 % in males versus 10.3 % in placebo group.
Neuroleptic malignant syndrome: A potentially fatal symptom complex referred to as Neuroleptic Malignant Syndrome (NMS) has been reported in association with brexpiprazole (see Precautions).
Nausea: For nausea, the incidence in the 2 mg to 4 mg brexpiprazole group was 2.3 % overall in short-term controlled trials, compared to 2.0 % in placebo; for vomiting, these incidences were 1.0 % in the brexpiprazole-treated group compared to 1.2 % in placebo group.
In terms of gender differences, there were higher observed frequencies of nausea (4.8 % versus 2.8 %) and vomiting (4.6 % versus 1.4 %) in females compared to males among brexpiprazole-treated subjects in short-term trials, in subjects receiving placebo: the frequency for nausea was 2.8 % for males versus 3.2 % for females and for vomiting the frequency was 3.0 % for males versus 2.6 % for females (see Pharmacology: Pharmacokinetics under Actions).
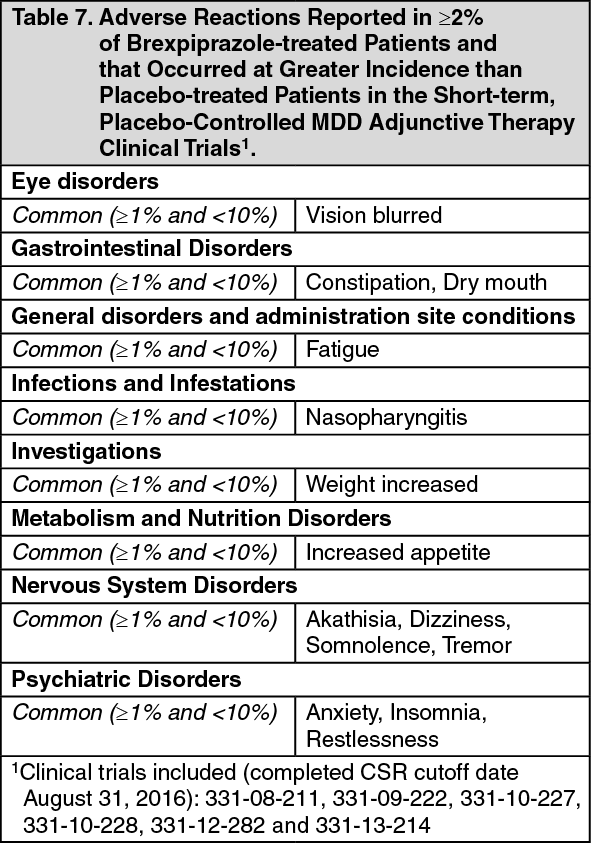
CLINICAL TRIAL DATA FOR MAJOR DEPRESSIVE DISORDER: Table 7 shows the incidence of adverse reactions that occurred in at least 2% of patients treated with 1-3 mg brexpiprazole+ADT treated group and observed more frequently than placebo. (See Table 7.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAdverse reactions that occurred <2% and the difference between brexpiprazole and placebo ≥0.5% in the short-term; placebo-controlled MDD adjunctive therapy clinical trials included palpitations, blepharospasm, toothache, salivary hypersecretion, urinary tract infection, blood prolactin increased, blood cortisol decreased, aspartate aminotransferase increased, muscle spasms, tension, night sweats and hypertension.
Selected Adverse Reactions: Extrapyramidal Symptoms: In the three 6-week, placebo-controlled, fixed-dose and one 6-week, placebo-controlled, flexible-dose MDD studies for brexpiprazole- treated patients, the incidence of reported EPS-related events, excluding akathisia events, was 5% versus 3% for placebo-treated patients. The incidence of akathisia events for brexpiprazole-treated patients was 8% versus 3% for placebo-treated patients.
Clinical Chemistry Findings: Weight Gain: In the long-term, open-label MDD studies, the mean change in body weight from baseline to last visit was 2.6 kg (N=2232). The proportion of patients with a ≥7% increase in body weight at last visit was 22.12% (494/2232) and with a ≥7% decrease in body weight was 3.2% (72/2232). At 52 weeks, the proportion of patients with a ≥7% increase in body weight was 28.2 % (286/1013) and with a ≥7% decrease in body weight was 3.7% (37/1013). Weight gain led to discontinuation of study medication in 3.8% (84/2240) of patients.
Fasting Glucose: In the long-term, open-label MDD studies, 5.22% of patients with normal baseline fasting glucose experienced a shift from normal to high while taking brexpiprazole, 24.35% of subjects with borderline fasting glucose experienced shifts from borderline to high. Combined, 9.06% of patients with normal or borderline fasting glucose experienced shifts to high fasting glucose during the long-term MDD studies. The mean change from baseline for fasting glucose to last visit in the long-term, open label trials was 3.53 [2.00] mg/dL.
Fasting Lipids: In the long-term open-label studies, shifts in baseline fasting cholesterol from normal to high were reported in 8.65% (total cholesterol), 3.20% (LDL cholesterol), and shifts in baseline from normal to low were reported in 13.30% (HDL cholesterol) of patients taking brexpiprazole. Of patients with normal baseline triglycerides, 17.26% experienced shifts to high, and 0.22% experienced shifts to very high triglycerides. Combined, 0.61% of patients with normal or borderline fasting triglycerides experienced shifts to very high fasting triglycerides during the long-term MDD studies. The mean changes from baseline for fasting HDL cholesterol, fasting LDL cholesterol, fasting cholesterol and fasting triglycerides to last visit in the long-term, open label trials were -2.13 [-2.00] mg/dL, 1.36 [1.00] mg/dL, 0.05 [0.00] mg/dL and 11.46 [8.00] mg/dL, respectively.
View ADR Monitoring Form