Pharmacotherapeutic group: Psycholeptics, other antipsychotics,
ATC code: N05AX16.
Pharmacology: Pharmacodynamics: Mechanism of action: Brexpiprazole is an atypical antipsychotic agent. The pharmacology of brexpiprazole is believed to be mediated by a modulatory activity at the serotonin and dopamine systems that combines partial agonist activity at serotonergic 5-HT
1A and at dopaminergic D
2 receptors with antagonist activity at serotonergic 5-HT
2A receptors, with similar high affinities at all of these receptors (Ki: 0.1 nM to 0.5 nM). Brexpiprazole also shows antagonist activity at noradrenergic α
1B/2C receptors with affinity in the same sub-nanomolar Ki range (Ki: 0.2 nM to 0.6 nM).
Pharmacodynamic effects: Influences of genetic variation on the pharmacodynamic responses to brexpiprazole have not been investigated.
Effects on QT: The effects of brexpiprazole on the QT interval were evaluated in patients with schizophrenia or schizoaffective disorder. In the overall analysis brexpiprazole did not prolong the QT
c interval to clinically relevant extent following therapeutic and supra-therapeutic doses (4 mg/day; n = 62 or 12 mg/day; n = 53) and no correlation has been observed between brexpiprazole concentrations and QT
c prolongation.
Subgroup analyses from the thorough QT
c trial suggested that the QT
c prolongation was larger in female subjects than in males. In the brexpiprazole 4 mg/day group, the maximum placebo-adjusted mean change from baseline in the QT
cl interval was 5.2 ms (90 % Cl: 1.5, 8.9) in males (n = 48) and 15.0 ms (90 % Cl: 7.7., 22.3) in females (n = 14) at 6 hours post-dosing. In the brexpiprazole 12 mg/day group, the maximum placebo-adjusted mean change from baseline in the QT
cl interval was 2.9 ms (90 % Cl: −1.2, 6.9) in males (n = 40) at 12 hours post-dosing and 10.4 ms (90 % Cl: 2.7, 18.2) in females (n = 13) at 24 hours post-dosing. The smaller number of female than male subjects enrolled in the study does not allow to draw definitive conclusions.
Clinical efficacy and safety: Schizophrenia: The efficacy and safety of brexpiprazole in the treatment of adults with schizophrenia was studied in two multi-national and one regional (Japan), 6-week, randomised, double-blind, placebo-controlled, fixed-dose clinical trials (trials 1 to 3), a multi-national, 6-week, randomised, double-blind, placebo-controlled, active reference (quetiapine), flexible-dose clinical trial (trial 4), and, one multi-national, placebo-controlled, 52-week maintenance trial (trial 5). The trials included 2,690 patients with the age of 18 years to 65 years.
In trials 1, 2 and 3 brexpiprazole was titrated as described in section 4.2 with 1 mg for 4 days, followed by 2 mg on days 5 to 7. On day 8 the dose was increased to 4 mg for some of the treatment arms.
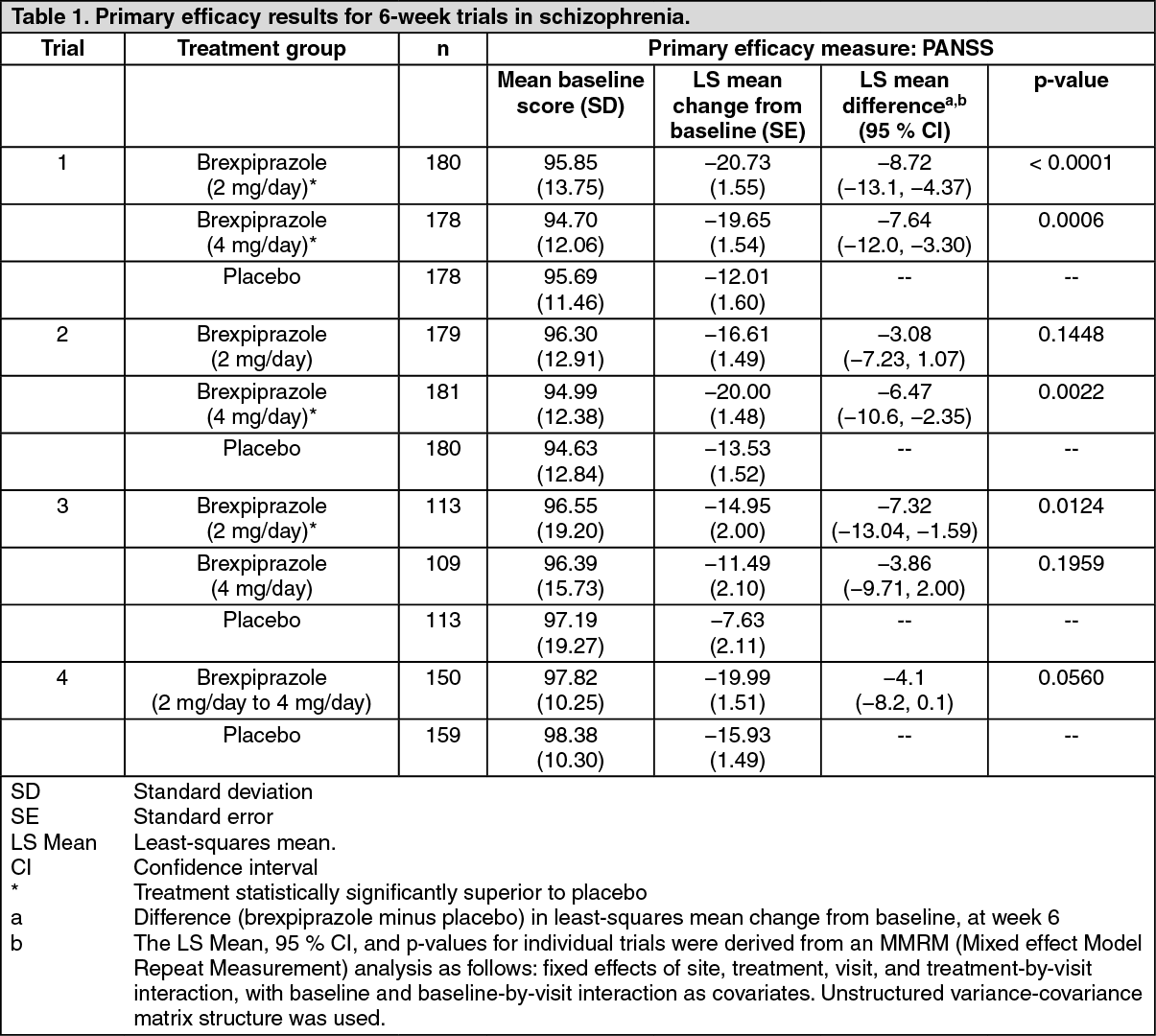
Short-term trials: In the three fixed-dose, short-term trials (trials 1, 2 and 3), subjects were randomised to brexpiprazole 2 mg once daily, 4 mg once daily or placebo.
Trial 4 assessed the efficacy, safety, and tolerability of brexpiprazole in a flexible dose range of 2 mg/day to 4 mg/day and 400 mg to 800 mg quetiapine extended release (XR) for assay sensitivity. In the short-term trials, the primary efficacy endpoint was defined as the mean change from baseline to week 6 in Positive and Negative Syndrome Scale (PANSS) total scores, a multi-item inventory composed of five factors to evaluate positive symptoms, negative symptoms, disorganised thoughts, uncontrolled hostility/excitement, and anxiety/depression.
The key secondary endpoint in trials 1, 2 and 4 was the Clinical Global Impression of Severity (CGI-S) of schizophrenia, a 7-point clinician's assessment of the severity of disease. The CGI-S was also assessed in trials 3 and 5 as secondary endpoint.
The effects of brexpiprazole were also evaluated across a number of pre-specified secondary endpoints; the specific aspects of symptoms of schizophrenia (PANSS Positive Subscale score, PANSS Negative Subscale score, PANSS Excited Component [PEC] score, PANSS Marder factors positive, negative, disorganised thoughts, uncontrolled hostility/excitement and anxiety/depression), and analyses of response (defined as 30 % improvement in PANSS total score compared to baseline or a CGI-I score of 1 [very much improved] or 2 [much improved]).
Efficacy was demonstrated in trial 1 for both brexpiprazole 2 mg/day and 4 mg/day and replicated in trial 2 only for brexpiprazole 4 mg/day and in trial 3 only for brexpiprazole 2 mg/day.
In the flexible-dose trial 4, at week 6, subjects in the brexpiprazole treatment group had numerically greater improvements on PANSS total score than the subjects in the placebo group, although, the difference at week 6 did not reach statistical significance for the primary efficacy analysis (p = 0.0560; see Table 1). In the same trial the active reference, quetiapine XR added for assay sensitivity only, separated from placebo. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The primary statistical analysis was performed using an MMRM model with MAR (Missing At Random) imputation. Results of a sensitivity analysis using placebo based multiple imputation (PMI) were consistent with the primary analysis.
Results for the (key) secondary outcome parameter and additional endpoints were supportive of the primary endpoint.
In trial 1, statistically significant greater improvement on the CGI-S, the key secondary efficacy measure, at week 6 was also shown for the 2 mg/day and 4 mg/day compared to the placebo dose groups. Due to the testing hierarchy the greater improvement shown for both 2 mg/day and 4 mg/day on the CGI-S can only be considered supportive for trials 2, 3 and 4 (see Table 2).
Click on icon to see table/diagram/image
Maintenance of efficacy trial: In trial 5, a long-term trial designed to assess the maintenance of effect of brexpiprazole by assessing the delay in time to impending relapse of schizophrenia, patients with schizophrenia, who responded to treatment with brexpiprazole 1 mg/day to 4 mg/day, were stabilised over 12 weeks to 36 weeks, and then randomised in a double-blind manner to either continue treatment with the stabilisation dose of brexpiprazole (n = 96) or to receive placebo (n = 104) for 52 weeks or until relapse occurred.
In the primary analysis of time to impending relapse patients on brexpiprazole showed a significantly longer time to relapse compared with patients on placebo (p < 0.0001). At week 52 brexpiprazole (13.5 %) reduced the risk of impending relapse by 71 % compared with placebo (38.5 %). During the stabilisation, brexpiprazole improved clinical symptomology (as assessed by PANSS, CGI-S and CGI-I, [Analysis of Covariance - ANCOVA Last Observation Carried Forward - LOCF]) and functioning (as assessed by Global Assessment of Functioning (GAF) [ANCOVA LOCF]). These improvements were maintained during the 52-week double-blind maintenance phase in patients on brexpiprazole whereas patients randomised to placebo showed deterioration in PANSS, CGI-S and CGI-I, and GAF scores [ANCOVA LOCF]). Brexpiprazole maintained symptom control and functioning compared to placebo.
Paediatric population: The European Medicines Agency has deferred the obligation to submit the results of efficacy and safety studies with brexpiprazole in the paediatric population from 13 years to less than 18 years of age (see Dosage & Administration for information on paediatric use).
Short-term Adjunctive Treatment in Major Depressive Disorder (MDD): The efficacy of , as an adjunctive treatment to antidepressant therapy for major depressive disorder (MDD), was evaluated in four phase 3, 6-week, double-blind, placebo-controlled trials: three fixed-dose trials (331-10-228, 331-10-227, 331-13-214) and one flexible-dose trial with an active reference (331-12-282). These trials are referred to as Trials 6, 7, 8 and 9, respectively, in Table 3.
The adult patients in these trials fulfilled the DSM-IV-TR criteria for MDD, with or without symptoms of anxiety, and demonstrated an inadequate response (patient reported) to 1-3 prior antidepressant therapy(ies) in the current episode and an inadequate response during the 8-10 weeks of prospective antidepressant treatment (escitalopram, fluoxetine, paroxetine controlled-release, sertraline, duloxetine or venlafaxine extended-release) during the trials. Inadequate response to prospective antidepressant treatment in Studies 6 and 7 was initially defined as < 50% improvement from baseline on the Hamilton Depression scale (HAMD-17), a HAMD-17 score > 14, and a Clinical Global Impression (CGI-I) ≥ 3 at Week 8. To ensure that randomized patients had an inadequate response throughout the prospective antidepressant treatment phase, this definition was amended during Studies 6 and 7 to the following: < 50% improvement from baseline on the HAMD-17 and a HAMD-17 score > 14 at Week 8; and, CGI-I ≥ 3 and < 50% improvement from baseline on the Montgomery-Asberg Depression Rating Scale (MADRS) Total Score at Weeks 2, 4, 6 and 8 (and Week 10, as applicable). This definition of inadequate response to prospective antidepressant treatment was also applied in Studies 8 and 9. With the exception of approximately 6% of patients in Studies 6 and 7, all patients who were randomized in the short-term clinical trials fulfilled the revised definition of inadequate response to prospective antidepressant treatment.
Patients remained on the same antidepressant treatment throughout the entire duration of each study. All patients randomized to in the fixed dose studies (Studies 6, 7, and 8) initiated treatment at 0.5 mg/day during Week 1. The dose was increased to 1 mg/day during Week 2 in all dose groups and, based on the assigned treatment, the dose was either maintained at 1 mg/day or increased to 3 mg/day (Study 7) or increased to 2 mg/day (Studies 6 and 8), from Week 3 onwards. Dosages were maintained at the assigned doses for the 4 remaining weeks. In the flexible dose study (Study 9), patients randomized to initiated treatment at 1 mg/day during Week 1, and the dose was increased to the target dose of 2 mg/day during Week 2. Patients remained at 2 mg/day in Study 9 unless there was a decision to increase the dose to 3 mg/day.
The primary efficacy endpoint in all studies was mean change from baseline (randomization) to Week 6 on the Montgomery Asberg Depression Rating Scale (MADRS) Total Score, a 10-item clinician-rated scale that assesses the degree of depressive symptomatology (apparent sadness, reported sadness, inner tension, reduced sleep, reduced appetite, concentration difficulties, lassitude, inability to feel, pessimistic thoughts, and suicidal thoughts). Each item is scored from 0 (normal/symptom not present) to 6 (most severe symptoms) and the range for the total score is 0 to 60.
The key secondary instrument was the Sheehan Disability Scale (SDS), a 3-item self-rated instrument used to assess three domains of functioning (work/school, social life, and family life) with each item scored from 0 (no disruption at all) to 10 (extreme disruption). (See Table 3.)
Click on icon to see table/diagram/image
Study Results: For the randomized patients, the mean duration of the current major depressive episode ranged between approximately 12 and 18 months and the majority of patients (approximately 79% - 84%) reported an inadequate response to one prior antidepressant treatment, before receiving 8-10 weeks of prospective antidepressant treatment during the trials. Following 8-10 weeks of prospective antidepressant treatment, the mean MADRS Total Score at randomization ranged between 25 and 27. Mean SDS score at randomization was between 5.6 and 6.3.
In Trials 6, 8 and 9 there was greater improvement in the mean MADRS Total Score with (2 mg/day or 2-3 mg/day) + ADT compared to placebo + ADT (p < 0.05). No additional benefit was demonstrated at doses greater than 2 mg/day (Table 4). In Study 9 the majority of patients treated with received 2 mg/day and the mean daily dose at endpoint was 2.2 mg/day. (See Table 4.)
Click on icon to see table/diagram/image
In Trial 6, the mean SDS score showed greater improvement with (2 mg/day) + ADT than with placebo + ADT (p<0.05).
Pharmacokinetics: Absorption: Brexpiprazole is absorbed after administration of the tablet, with peak plasma concentrations occurring within 4.0 hours after single dose administrations; the absolute oral bioavailability of the tablet formulation is 95.1 %. Brexpiprazole steady-state concentrations are attained within 10 days to 12 days of dosing. Administration of a 4 mg brexpiprazole tablet with a standard high fat meal did not significantly affect the C
max or AUC of brexpiprazole. After single and multiple once daily dose administration, brexpiprazole exposure (C
max and AUC) increase in proportion to the dose administered. Based on
in vivo studies, brexpiprazole is neither a substrate nor an inhibitor of efflux transporters, such as Multi Drug Resistance (MDR) 1 (P-gp) and BCRP.
Distribution: The volume of distribution of brexpiprazole following intravenous administration is high (1.56 L/kg ± 0.418 L/kg), indicating extravascular distribution. Brexpiprazole is highly protein bound in plasma (greater than 99 %) to serum albumin and α1-acid glycoprotein, and its protein binding is not affected by renal or hepatic impairment. Based on results of
in vitro studies brexpiprazole protein binding is not affected by warfarin, diazepam, and digitoxin.
Biotransformation: Based on
in vitro metabolism studies using recombinant human cytochrome P450, the metabolism of brexpiprazole was shown to be mainly mediated by CYP3A4 and CYP2D6 leading to formation of oxidative metabolites. Based on
in vitro data brexpiprazole showed little to no inhibition of other CYP450 isozymes.
In vivo, the metabolism of brexpiprazole is mainly mediated by CYP3A4 and CYP2D6 leading to formation of oxidative metabolites with only one metabolite, DM-3411, present in plasma with more than 10 % of plasma exposure.
At steady-state, DM-3411 represents 23.1 % to 47.7 % of brexpiprazole exposure (AUC) in plasma. It should be noted that
in vivo preclinical studies have shown that at clinically relevant plasma exposures of brexpiprazole, DM-3411 brain exposures were below the detection limit. Thus, DM-3411 is considered not to contribute to the therapeutic effects of brexpiprazole.
Elimination: Following a single oral dose of [
14C]-labelled brexpiprazole, approximately 24.6 % and 46 % of the administered radioactivity was recovered in the urine and faeces, respectively. Less than 1 % of unchanged brexpiprazole was excreted in the urine and approximately 14 % of the oral dose was recovered unchanged in the faeces. Apparent oral clearance of brexpiprazole tablet after once daily administration is 19.8 (± 11.4) mL/h/kg. After multiple once daily administration of brexpiprazole, the terminal elimination half-life of brexpiprazole and its major metabolite, DM-3411, is 91.4 hours and 85.7 hours, respectively.
Linearity/non-linearity: The pharmacokinetic of brexpiprazole is dose proportional and time-invariant after single-dose (0.2 mg to 8 mg) and multiple-dose (0.5 mg to 4 mg) using once-daily administration.
Pharmacokinetics in special populations: Age: After single dose administration of brexpiprazole (2 mg), elderly subjects (older than 65 years) exhibited similar brexpiprazole systemic exposure (C
max and AUC) in comparison with the adult subjects (18 years to 45 years old; see Dosage & Administration and Precautions).
Gender: Population PK evaluation identified gender as statistically significant covariate. The exposure (AUC) of brexpiprazole in women was estimated to be 25 % higher than in men (see Adverse Reactions).
Race: Although no specific pharmacokinetic study was conducted, population pharmacokinetic evaluation revealed no evidence of clinically significant race-related differences in the pharmacokinetics of brexpiprazole.
CYP2D6 genotype: Population pharmacokinetic evaluation shows that CYP2D6 poor metabolisers have 47 % higher exposure to brexpiprazole compared to extensive metabolisers (see Dosage & Administration).
Smoking: Based on studies utilising human liver enzymes
in vitro, brexpiprazole is not a substrate for CYP1A2; smoking should, therefore, not have an effect on the pharmacokinetics of brexpiprazole.
Renal impairment: In subjects (n = 10) with severe renal impairment (CL
cr < 30 mL/min), AUC of oral brexpiprazole (3 mg single dose) compared to matched healthy subjects was increased by 68 % while its C
max was not changed. For patients with moderate to severe renal impairment (creatinine clearance CL
cr < 60 mL/minute), the maximum recommended dose is reduced to 3 mg once daily for patients with schizophrenia and 2 mg once daily for patients with MDD (see Dosage & Administration).
Hepatic impairment: In subjects (n = 22) with varying degrees of hepatic impairment (Child-Pugh Classes A, B, and C), the AUC of oral brexpiprazole (2 mg single dose), compared to matched healthy subjects, increased 24 % in mild hepatic impairment, increased 60 % in moderate hepatic impairment, and did not change in severe hepatic impairment. For patients with moderate to severe hepatic impairment (Child-Pugh Classes B and C), the maximum recommended dose is reduced to 3 mg once daily for patients with schizophrenia and 2 mg once daily for patients with MDD (see Dosage & Administration).
Paediatric population: The safety and efficacy of brexpiprazole in children and adolescents aged less than 18 years have not been established (see Dosage & Administration).
Toxicology: Preclinical safety data: Effects observed in repeated-dose toxicity studies in rats and monkeys were mainly related to the exaggerated pharmacological activity of brexpiprazole. No safety margins based on AUC
0-24 h at the Maximum Recommended Human Dose (MRHD) of 4 mg/day could be derived in both female and male rats and monkey.
Cardiovascular toxicity: Following oral administration, brexpiprazole decreased blood pressure and prolonged QT interval in safety pharmacology study in conscious male dog, in repeated-dose toxicity studies in male and female monkeys and in juvenile toxicity study in male and female dogs. The effect of brexpiprazole on blood pressure reduction is attributed to the expected blockade of α1-adrenoceptors in peripheral blood vessels.
Genotoxicity, carcinogenicity: Brexpiprazole did not show any genotoxic potential in both
in vitro and
in vivo studies using clinically relevant exposures. Brexpiprazole administered orally did not increase in the incidence of tumours in 2-year carcinogenicity study in both male and female rats and in male mice at exposures up to 4.4-fold and 3.1-fold the MRHD. In female mice, an increased incidence of mammary gland adenocarcinoma and adeno-squamous carcinoma, and pars distalis adenoma of the pituitary gland, was observed at similar or even lower clinically relevant exposures: these prolactin-mediated endocrine tumours were also observed in rodents with other antipsychotics and their clinical relevance is unknown.
Reproductive toxicity: Following oral administration, brexpiprazole did not affect male fertility in rats but prolonged diestrus and decreased fertility in female rats at similar or even lower exposure levels than those clinically achieved at MRHD. Significant increased pre-implantation losses were observed at 4.1-fold the clinical exposure at MRHD. In embryo-foetal developmental toxicity studies, brexpiprazole was not teratogen in orally treated rats up to exposure levels (based on data in non-pregnant rats) clinically achieved at MRHD. In rabbit, vertebral malformations were seen in 3 foetuses from 2 litters at 21 maternally toxic brexpiprazole oral doses corresponding to exposure approximately 16.5-fold the clinical exposure at MRHD.
Delayed growth, physical development and impaired viability of the offspring were observed at maternally toxic brexpiprazole doses in a pre-/post-natal developmental toxicity study in orally administered rats.
Following oral administration in pregnant rats, foetus and milk transfer of brexpiprazole was demonstrated at concentrations that were generally comparable to levels seen in maternal blood.
Environmental risk assessment (ERA): Brexpiprazole is very persistent and very bioaccumulative but not toxic, to the environment: possible enrichment of brexpiprazole in terrestrial food chains might pose a concern (see Cautions for Usage).


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image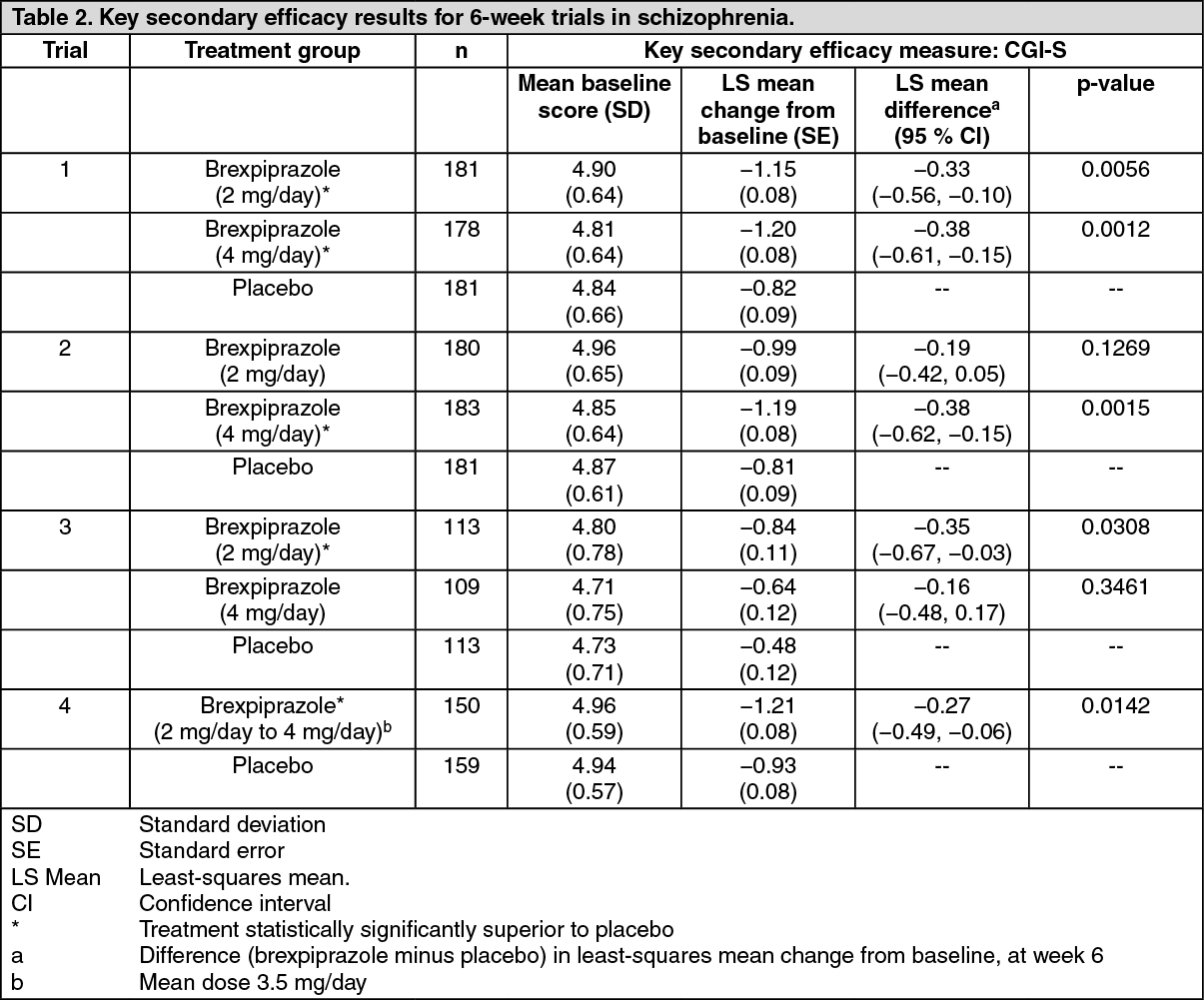 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image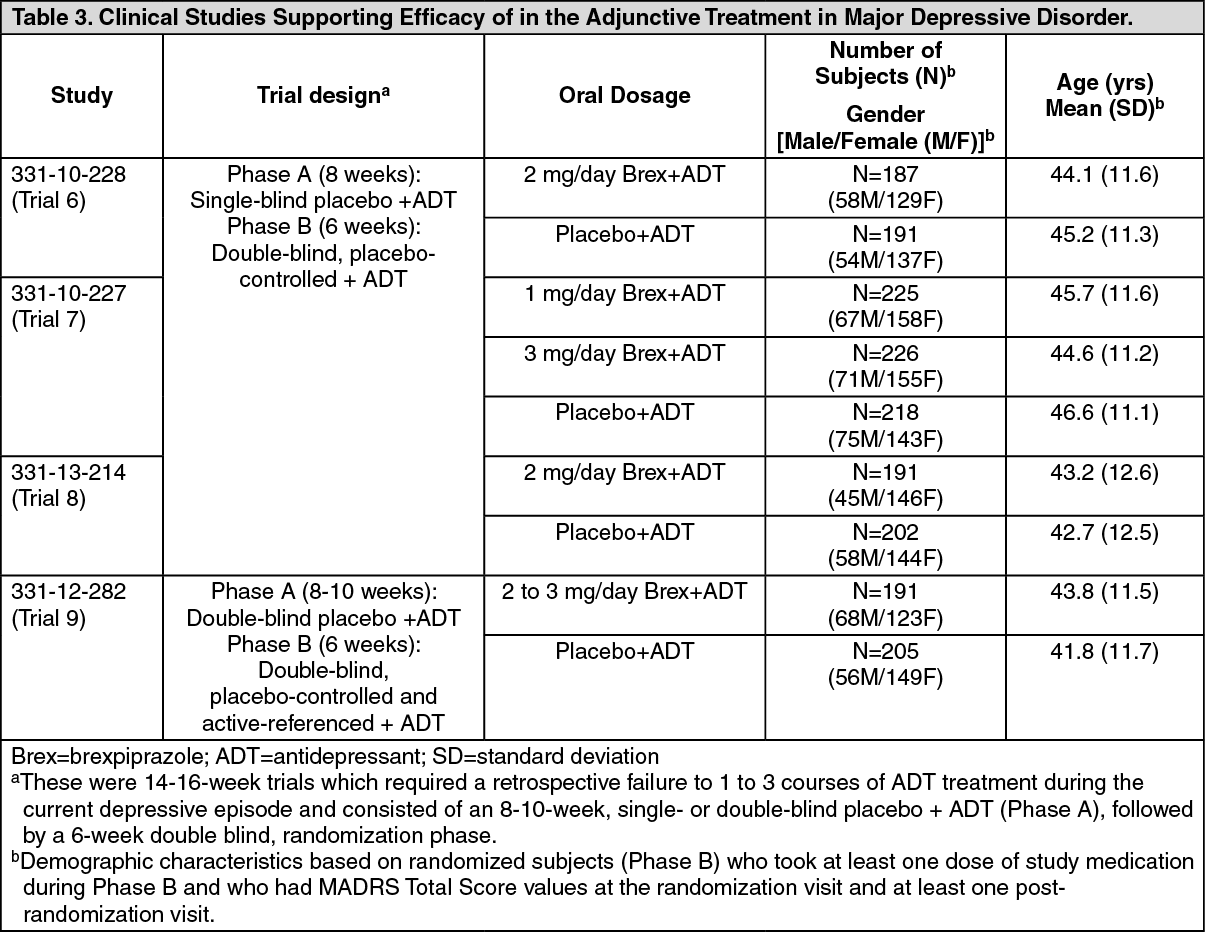 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image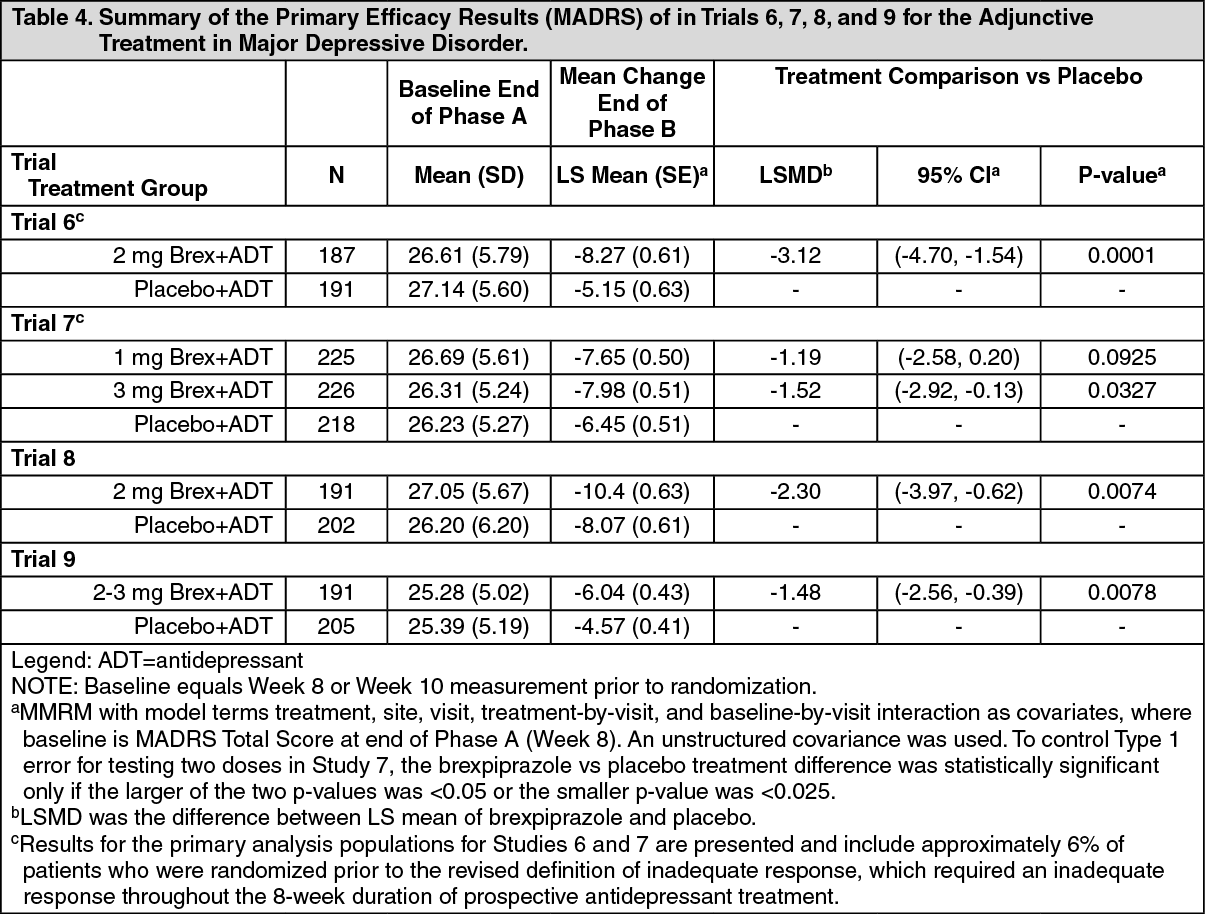 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image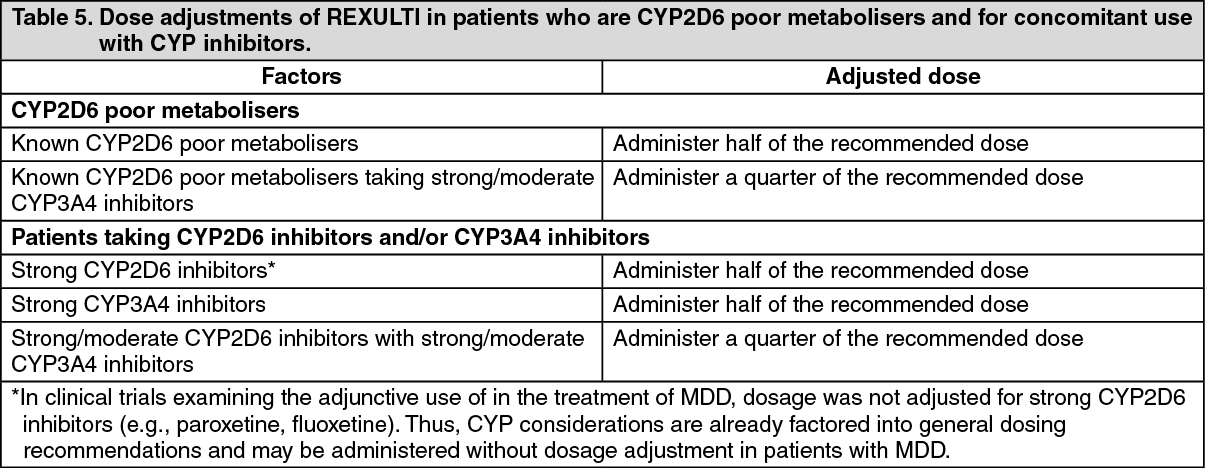 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image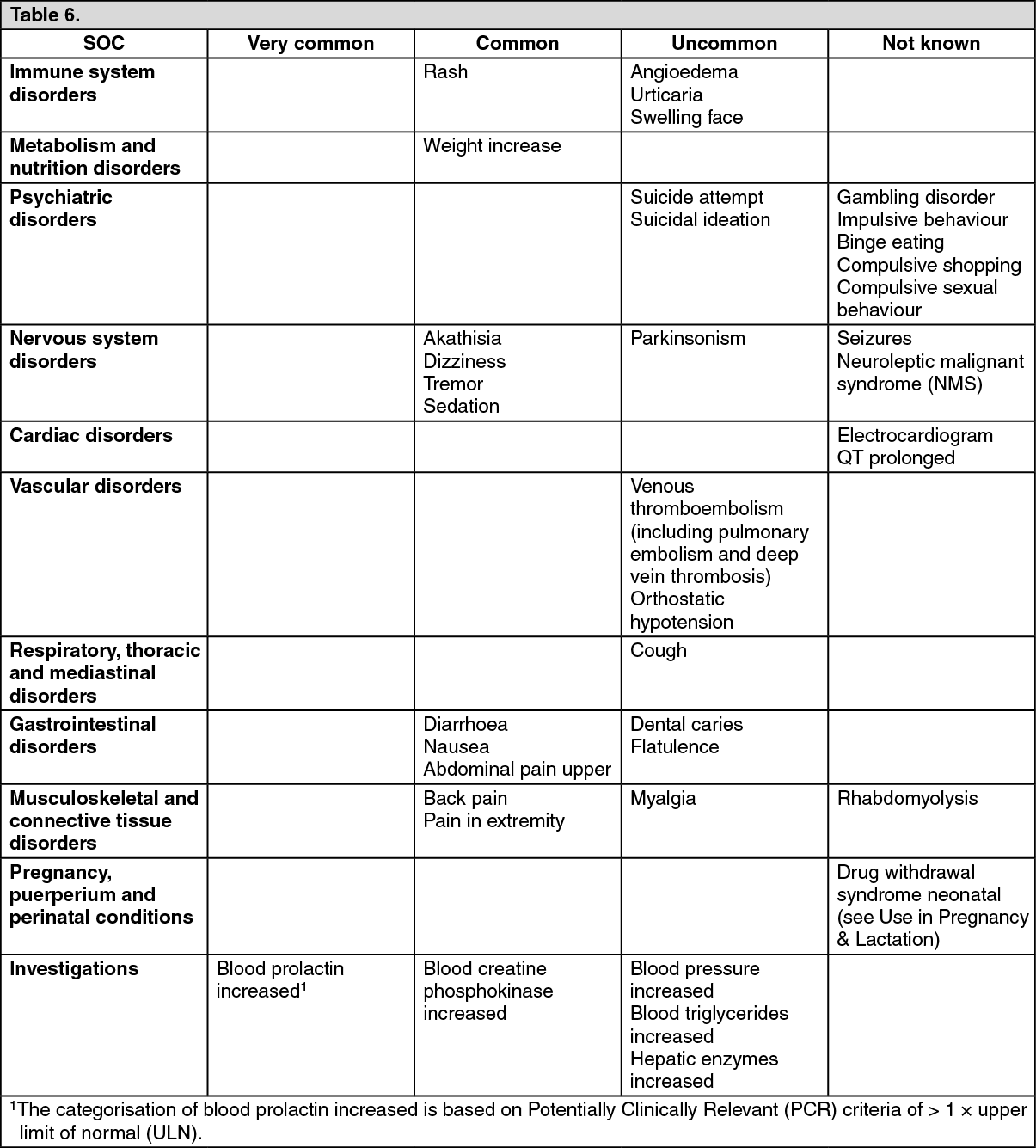 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image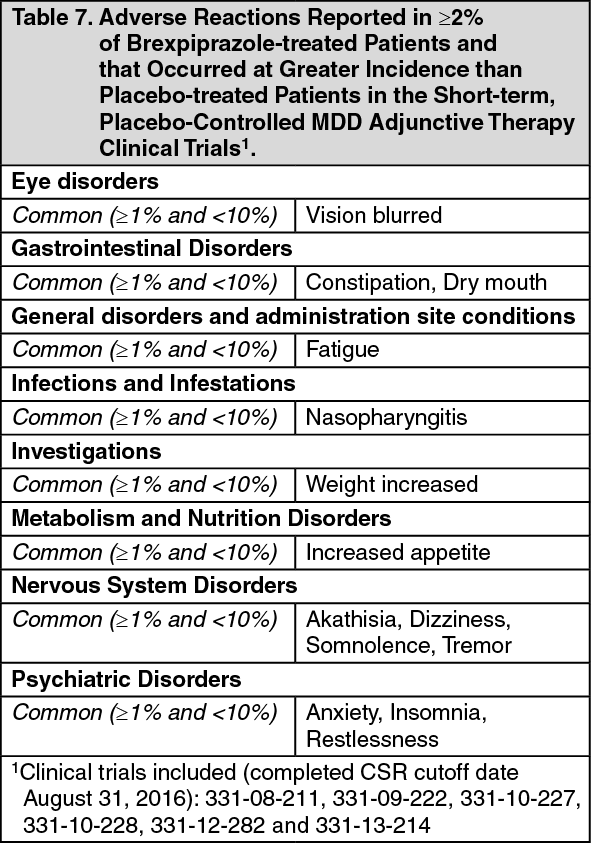 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image


 Sign Out
Sign Out
