


 Sign Out
Sign Out

The most frequently observed adverse drug reactions (ADRs) in patients receiving rituximab were IRRs which occurred in the majority of patients during the first infusion. The incidence of infusion-related symptoms decreases substantially with subsequent infusions and is less than 1% after eight doses of rituximab.
Infectious events (predominantly bacterial and viral) occurred in approximately 30-55% of patients during clinical trials in patients with NHL and in 30-50% of patients during clinical trials in patients with CLL.
The most frequent reported or observed serious adverse drug reactions were: IRRs (including cytokine-release syndrome, tumour-lysis syndrome), see Precautions.
Infections, see Precautions.
Cardiovascular events, see Precautions.
Other serious ADRs reported include hepatitis B reactivation and PML (see Precautions).
List of adverse reactions: The frequencies of ADRs reported with rituximab alone or in combination with chemotherapy are summarised as follows. Within each frequency grouping, undesirable effects are presented in order of decreasing seriousness. Frequencies are defined as very common (≥1/10), common (≥1/100 to <1/10), uncommon (≥1/1,000 to <1/100), rare (≥1/10,000 to <1/1000), very rare (<1/10,000) and not known (cannot be estimated from the available data).
The ADRs identified only during post-marketing surveillance, and for which a frequency could not be estimated, are listed under "not known". (See Table 14.)
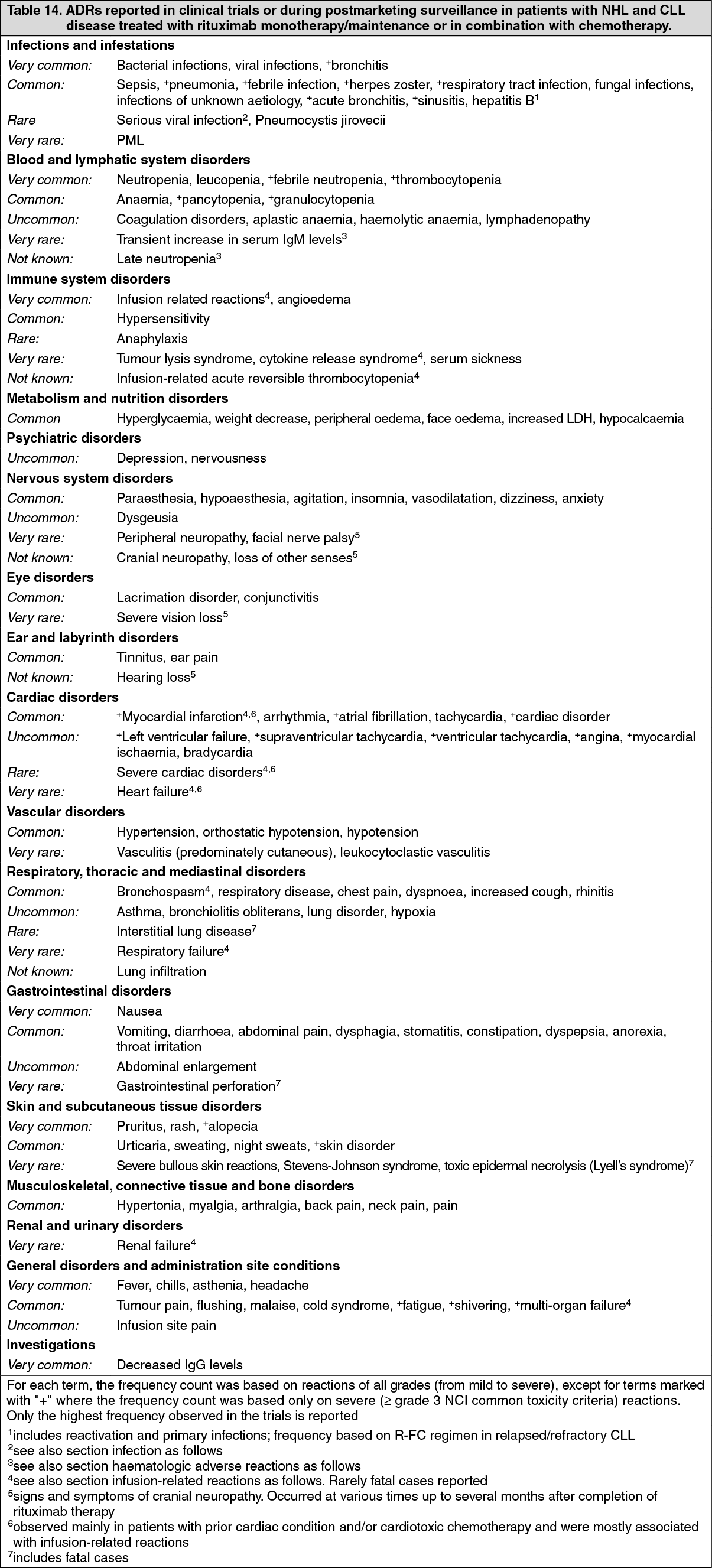 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe following terms have been reported as adverse events during clinical trials, however, were reported at a similar or lower incidence in the rituximab arms compared to control arms: haematotoxicity, neutropenic infection, urinary tract infection, sensory disturbance, pyrexia.
Signs and symptoms suggestive of an infusion-related reaction were reported in more than 50% of patients in clinical trials, and were predominantly seen during the first infusion, usually in the first one to two hours. These symptoms mainly comprised fever, chills and rigors. Other symptoms included flushing, angioedema, bronchospasm, vomiting, nausea, urticaria/rash, fatigue, headache, throat irritation, rhinitis, pruritus, pain, tachycardia, hypertension, hypotension, dyspnoea, dyspepsia, asthenia and features of tumour lysis syndrome. Severe infusion-related reactions (such as bronchospasm, hypotension) occurred in up to 12% of the cases. Additional reactions reported in some cases were myocardial infarction, atrial fibrillation, pulmonary oedema and acute reversible thrombocytopenia. Exacerbations of pre-existing cardiac conditions such as angina pectoris or congestive heart failure or severe cardiac disorders (heart failure, myocardial infarction, atrial fibrillation), pulmonary oedema, multi-organ failure, tumour lysis syndrome, cytokine release syndrome, renal failure, and respiratory failure were reported at lower or unknown frequencies. The incidence of infusion-related symptoms decreased substantially with subsequent infusions and is <1% of patients by the eighth cycle of rituximab (containing) treatment.
Description of selected adverse reactions: Infections: Rituximab induces B-cell depletion in about 70-80% of patients, but was associated with decreased serum immunoglobulins only in a minority of patients.
Localised candida infections as well as Herpes zoster were reported at a higher incidence in the rituximab-containing arm of randomised studies. Severe infections were reported in about 4% of patients treated with rituximab monotherapy. Higher frequencies of infections overall, including grade 3 or 4 infections, were observed during rituximab maintenance treatment up to 2 years when compared to observation. There was no cumulative toxicity in terms of infections reported over a 2-year treatment period. In addition, other serious viral infections either new, reactivated or exacerbated, some of which were fatal, have been reported with rituximab treatment. The majority of patients had received rituximab in combination with chemotherapy or as part of a haematopoetic stem cell transplant. Examples of these serious viral infections are infections caused by the herpes viruses (Cytomegalovirus, Varicella Zoster Virus and Herpes Simplex Virus), JC virus (progressive multifocal leukoencephalopathy (PML)) and hepatitis C virus. Cases of fatal PML that occurred after disease progression and retreatment have also been reported in clinical trials. Cases of hepatitis B reactivation, have been reported, the majority of which were in patients receiving rituximab in combination with cytotoxic chemotherapy. In patients with relapsed/refractory CLL, the incidence of grade 3/4 hepatitis B infection (reactivation and primary infection) was 2% in R-FC vs 0% FC. Progression of Kaposi's sarcoma has been observed in rituximab-exposed patients with pre-existing Kaposi's sarcoma. These cases occurred in non-approved indications and the majority of patients were HIV positive.
Haematologic adverse reactions: In clinical trials with rituximab monotherapy given for 4 weeks, haematological abnormalities occurred in a minority of patients and were usually mild and reversible. Severe (grade 3/4) neutropenia was reported in 4.2%, anaemia in 1.1% and thrombocytopenia in 1.7% of the patients. During rituximab maintenance treatment for up to 2 years, leucopenia (5% vs. 2%, grade 3/4) and neutropenia (10% vs. 4%, grade 3/4) were reported at a higher incidence when compared to observation. The incidence of thrombocytopenia was low (<1%, grade 3/4%) and was not different between treatment arms. During the treatment course in studies with rituximab in combination with chemotherapy, grade 3/4 leucopenia (R-CHOP 88% vs. CHOP 79%, R-FC 23% vs. FC 12%), neutropenia (R-CVP 24% vs. CVP 14%; R-CHOP 97% vs. CHOP 88%, R-FC 30% vs. FC 19% in previously untreated CLL), pancytopenia (R-FC 3% vs. FC 1% in previously untreated CLL) were usually reported with higher frequencies when compared to chemotherapy alone. However, the higher incidence of neutropenia in patients treated with rituximab and chemotherapy was not associated with a higher incidence of infections and infestations compared to patients treated with chemotherapy alone. Studies in previously untreated and relapsed/refractory CLL have established that in up to 25% of patients treated with R-FC neutropenia was prolonged (defined as neutrophil count remaining below 1x109/l between day 24 and 42 after the last dose) or occurred with a late onset (defined as neutrophil count below 1x109/l later than 42 days after last dose in patients with no previous prolonged neutropenia or who recovered prior to day 42) following treatment with rituximab plus FC. There were no differences reported for the incidence of anaemia. Some cases of late neutropenia occurring more than four weeks after the last infusion of rituximab were reported. In the CLL first-line study, Binet stage C patients experienced more adverse events in the R -FC arm compared to the FC arm (R-FC 83% vs. FC 71%). In the relapsed/refractory CLL study grade ¾ thrombocytopenia was reported in 11% of patients in the R-FC group compared to 9% of patients in the FC group.
In studies of rituximab in patients with Waldenstrom's macroglobulinaemia, transient increases in serum IgM levels have been observed following treatment initiation, which may be associated with hyperviscosity and related symptoms. The transient IgM increase usually returned to at least baseline level within 4 months.
Cardiovascular adverse reactions: Cardiovascular reactions during clinical trials with rituximab monotherapy were reported in 18.8% of patients with the most frequently reported events being hypotension and hypertension. Cases of grade 3 or 4 arrhythmia (including ventricular and supraventricular tachycardia) and angina pectoris during infusion were reported. During maintenance treatment, the incidence of grade 3/4 cardiac disorders was comparable between patients treated with rituximab and observation. Cardiac events were reported as serious adverse events (including atrial fibrillation, myocardial infarction, left ventricular failure, myocardial ischaemia) in 3% of patients treated with rituximab compared to <1% on observation. In studies evaluating rituximab in combination with chemotherapy, the incidence of grade 3 and 4 cardiac arrhythmias, predominantly supraventricular arrhythmias such as tachycardia and atrial flutter/fibrillation, was higher in the R-CHOP group (14 patients, 6.9%) as compared to the CHOP group (3 patients, 1.5%). All of these arrhythmias either occurred in the context of a rituximab infusion or were associated with predisposing conditions such as fever, infection, acute myocardial infarction or pre-existing respiratory and cardiovascular disease. No difference between the R-CHOP and CHOP group was observed in the incidence of other grade 3 and 4 cardiac events including heart failure, myocardial disease and manifestations of coronary artery disease. In CLL, the overall incidence of grade 3 or 4 cardiac disorders was low both in the first-line study (4% R-FC, 3% FC) and in the relapsed/refractory study (4% R-FC, 4% FC).
Respiratory system: Cases of interstitial lung disease, some with fatal outcome have been reported.
Neurologic disorders: During the treatment period (induction treatment phase comprising of R-CHOP for at most eight cycles), four patients (2%) treated with R-CHOP, all with cardiovascular risk factors, experienced thromboembolic cerebrovascular accidents during the first treatment cycle. There was no difference between the treatment groups in the incidence of other thromboembolic events. In contrast, three patients (1.5%) had cerebrovascular events in the CHOP group, all of which occurred during the follow-up period. In CLL, the overall incidence of grade 3 or 4 nervous system disorders was low both in the first-line study (4% R-FC, 4% FC) and in the relapsed/refractory study (3% R-FC, 3% FC).
Cases of posterior reversible encephalopathy syndrome (PRES) / reversible posterior leukoencephalopathy syndrome (RPLS) have been reported. Signs and symptoms included visual disturbance, headache, seizures and altered mental status, with or without associated hypertension. A diagnosis of PRES/RPLS requires confirmation by brain imaging. The reported cases had recognised risk factors for PRES/RPLS, including the patients' underlying disease, hypertension, immunosuppressive therapy and/or chemotherapy.
Gastrointestinal disorders: Gastrointestinal perforation in some cases leading to death has been observed in patients receiving rituximab for treatment of non-Hodgkin lymphoma. In the majority of these cases, rituximab was administered with chemotherapy.
IgG levels: In the clinical trial evaluating rituximab maintenance treatment in relapsed/refractory follicular lymphoma, median IgG levels were below the lower limit of normal (LLN) (<7 g/l) after induction treatment in both the observation and the rituximab groups. In the observation group, the median IgG level subsequently increased to above the LLN, but remained constant in the rituximab group. The proportion of patients with IgG levels below the LLN was about 60% in the rituximab group throughout the 2 year treatment period, while it decreased in the observation group (36% after 2 years).
A small number of spontaneous and literature cases of hypogammaglobulinaemia have been observed in paediatric patients treated with rituximab, in some cases severe and requiring long-term immunoglobulin substitution therapy. The consequences of long term B cell depletion in paediatric patients are unknown.
Skin and subcutaneous tissue disorders: Toxic epidermal necrolysis (Lyell syndrome) and Stevens-Johnson syndrome, some with fatal outcome, have been reported very rarely.
Patient subpopulations - rituximab monotherapy: Elderly patients (≥65 years): The incidence of ADRs of all grades and grade 3/4 ADR was similar in elderly patients compared to younger patients (<65 years).
Bulky disease: There was a higher incidence of grade 3/4 ADRs in patients with bulky disease than in patients without bulky disease (25.6% vs. 15.4%). The incidence of ADRs of any grade was similar in these two groups.
Re-treatment: The percentage of patients reporting ADRs upon re-treatment with further courses of rituximab was similar to the percentage of patients reporting ADRs upon initial exposure (any grade and grade ¾ ADRs).
Patient subpopulations - rituximab combination therapy: Elderly patients (≥65 years): The incidence of grade 3/4 blood and lymphatic adverse events was higher in elderly patients compared to younger patients (<65 years), with previously untreated or relapsed/refractory CLL.
Experience from rheumatoid arthritis: Summary of the safety profile: The overall safety profile of rituximab in rheumatoid arthritis is based on data from patients from clinical trials and from post-marketing surveillance.
The safety profile of rituximab in patients with moderate to severe rheumatoid arthritis (RA) is summarised in the sections as follows. In clinical trials more than 3100 patients received at least one treatment course and were followed for periods ranging from 6 months to over 5 years; approximately 2400 patients received two or more courses of treatment with over 1000 having received 5 or more courses. The safety information collected during post-marketing experience reflects the expected adverse reaction profile as seen in clinical trials for rituximab (see Precautions).
Patients received 2 x 1000 mg of rituximab separated by an interval of two weeks; in addition to methotrexate (10-25 mg/week). Rituximab infusions were administered after an intravenous infusion of 100 mg methylprednisolone; patients also received treatment with oral prednisone for 15 days.
List of adverse reactions: Adverse reactions are listed as follows. Frequencies are defined as very common (≥1/10), common (≥1/100 to <1/10), uncommon (≥1/1,000 to <1/100), and very rare (<1/10,000). Within each frequency grouping, undesirable effects are presented in order of decreasing seriousness.
The most frequent adverse reactions considered due to receipt of rituximab were IRRs. The overall incidence of IRRs in clinical trials was 23% with the first infusion and decreased with subsequent infusions. Serious IRRs were uncommon (0.5% of patients) and were predominantly seen during the initial course. In addition to adverse reactions seen in RA clinical trials for rituximab, progressive multifocal leukoencephalopathy (PML) (see Precautions) and serum sickness-like reaction have been reported during post marketing experience. (See Table 15.)
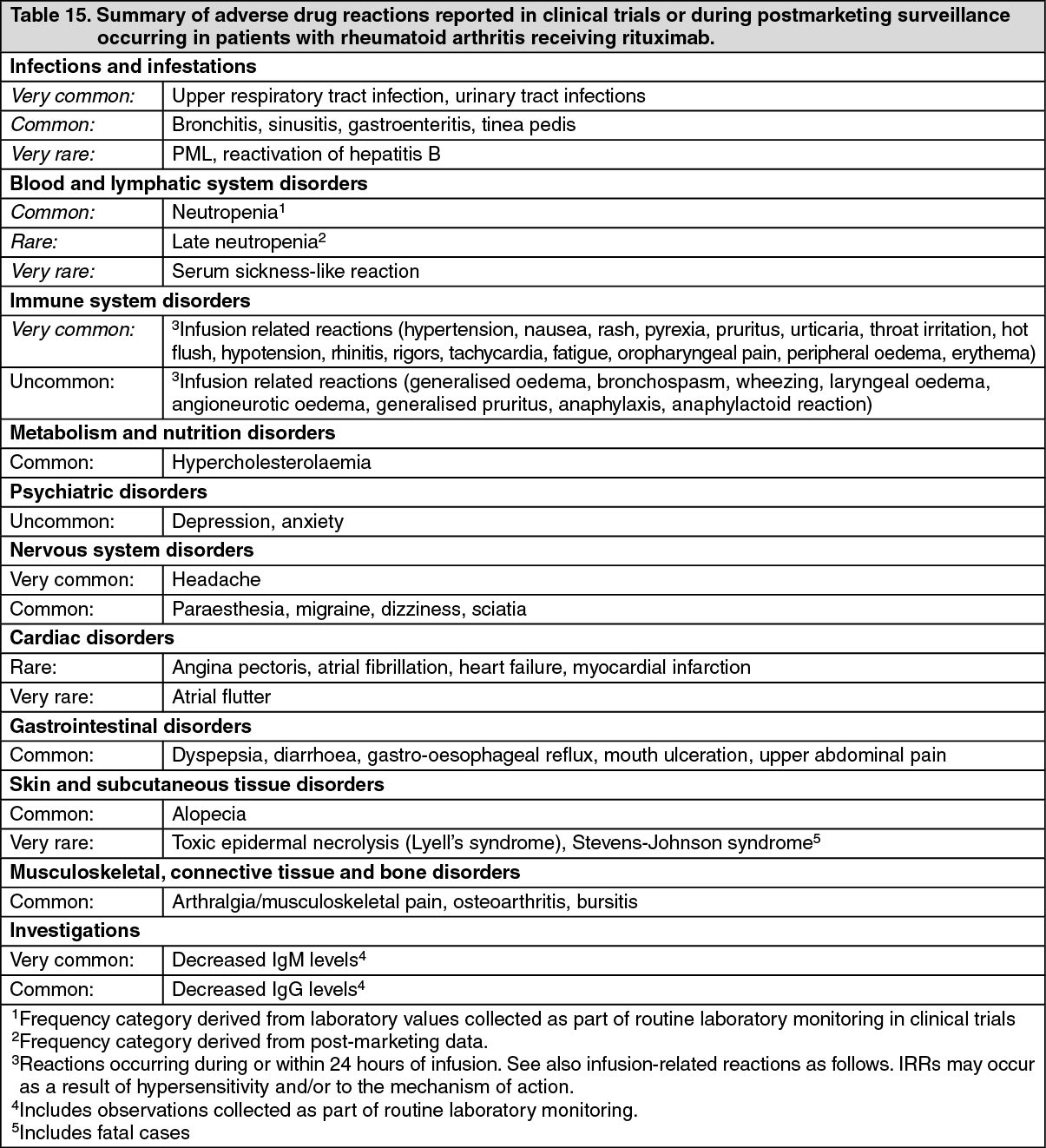 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageMultiple courses: Multiple courses of treatment are associated with a similar ADR profile to that observed following first exposure. The rate of all ADRs following first rituximab exposure was highest during the first 6 months and declined thereafter. This is mostly accounted for by IRRs (most frequent during the first treatment course), RA exacerbation and infections, all of which were more frequent in the first 6 months of treatment.
Infusion-related reactions: The most frequent ADRs following receipt of rituximab in clinical studies were IRRs (see in the previous text). Among the 3189 patients treated with rituximab, 1135 (36%) experienced at least one IRR with 733/3189 (23%) of patients experiencing an IRR following first infusion of the first exposure to rituximab. The incidence of IRRs declined with subsequent infusions. In clinical trials fewer than 1% (17/3189) of patients experienced a serious IRR. There were no CTC Grade 4 IRRs and no deaths due to IRRs in the clinical trials. The proportion of CTC Grade 3 events and of IRRs leading to withdrawal decreased by course and were rare from course 3 onwards. Premedication with intravenous glucocorticoid significantly reduced the incidence and severity of IRRs (see Dosage & Administration and Precautions). Severe IRRs with fatal outcome have been reported in the post-marketing setting.
In a trial designed to evaluate the safety of a more rapid rituximab infusion in patients with rheumatoid arthritis, patients with moderate-to-severe active RA who did not experience a serious IRR during or within 24 hours of their first studied infusion were allowed to receive a 2-hour intravenous infusion of rituximab. Patients with a history of a serious infusion reaction to a biologic therapy for RA were excluded from entry. The incidence, types and severity of IRRs were consistent with that observed historically. No serious IRRs were observed.
Description of selected adverse reactions: Infections: The overall rate of infection was approximately 94 per 100 patient years in rituximab treated patients. The infections were predominately mild to moderate and consisted mostly of upper respiratory tract infections and urinary tract infections. The incidence of infections that were serious or required IV antibiotics was approximately 4 per 100 patient years. The rate of serious infections did not show any significant increase following multiple courses of rituximab. Lower respiratory tract infections (including pneumonia) have been reported during clinical trials, at a similar incidence in the rituximab arms compared to control arms.
Cases of progressive multifocal leukoencephalopathy with fatal outcome have been reported following use of rituximab for the treatment of autoimmune diseases. This includes rheumatoid arthritis and off-label autoimmune diseases, including Systemic Lupus Erythematosus (SLE) and vasculitis.
In patients with non-Hodgkin's lymphoma receiving rituximab in combination with cytotoxic chemotherapy, cases of hepatitis B reactivation have been reported (see non-Hodgkin's lymphoma). Reactivation of hepatitis B infection has also been very rarely reported in RA patients receiving rituximab (see Precautions).
Cardiovascular adverse reactions: Serious cardiac reactions were reported at a rate of 1.3 per 100 patient years in the rituximab treated patients compared to 1.3 per 100 patient years in placebo treated patients. The proportions of patients experiencing cardiac reactions (all or serious) did not increase over multiple courses.
Neurologic events: Cases of posterior reversible encephalopathy syndrome (PRES) / reversible posterior leukoencephalopathy syndrome (RPLS) have been reported. Signs and symptoms included visual disturbance, headache, seizures and altered mental status, with or without associated hypertension. A diagnosis of PRES/RPLS requires confirmation by brain imaging. The reported cases had recognised risk factors for PRES/RPLS, including the patients' underlying disease, hypertension, immunosuppressive therapy and/or chemotherapy.
Neutropenia: Events of neutropenia were observed with rituximab treatment, the majority of which were transient and mild or moderate in severity. Neutropenia can occur several months after the administration of rituximab (see Precautions).
In placebo-controlled periods of clinical trials, 0.94% (13/1382) of rituximab treated patients and 0.27% (2/731) of placebo patients developed severe neutropenia.
Neutropenic events, including severe late onset and persistent neutropenia, have been rarely reported in the post-marketing setting, some of which were associated with fatal infections.
Skin and subcutaneous tissue disorders: Toxic epidermal necrolysis (Lyell's syndrome) and Stevens-Johnson syndrome, some with fatal outcome, have been reported very rarely.
Laboratory abnormalities: Hypogammaglobulinaemia (IgG or IgM below the lower limit of normal) has been observed in RA patients treated with rituximab. There was no increased rate in overall infections or serious infections after the development of low IgG or IgM (see Precautions).
A small number of spontaneous and literature cases of hypogammaglobulinaemia have been observed in paediatric patients treated with rituximab, in some cases severe and requiring long-term immunoglobulin substitution therapy. The consequences of long term B cell depletion in paediatric patients are unknown.
Experience from granulomatosis with polyangiitis and microscopic polyangiitis: In the clinical trial in granulomatosis with polyangiitis and microscopic polyangitis, 99 patients were treated with rituximab (375 mg/m2, once weekly for 4 weeks) and glucocorticoids (see Pharmacology: Pharmacodynamics under Actions).
Tabulated list of adverse reactions: The ADRs listed in Table 16 were all adverse events which occurred at an incidence of ≥5% in the rituximab group. (See Table 16.)
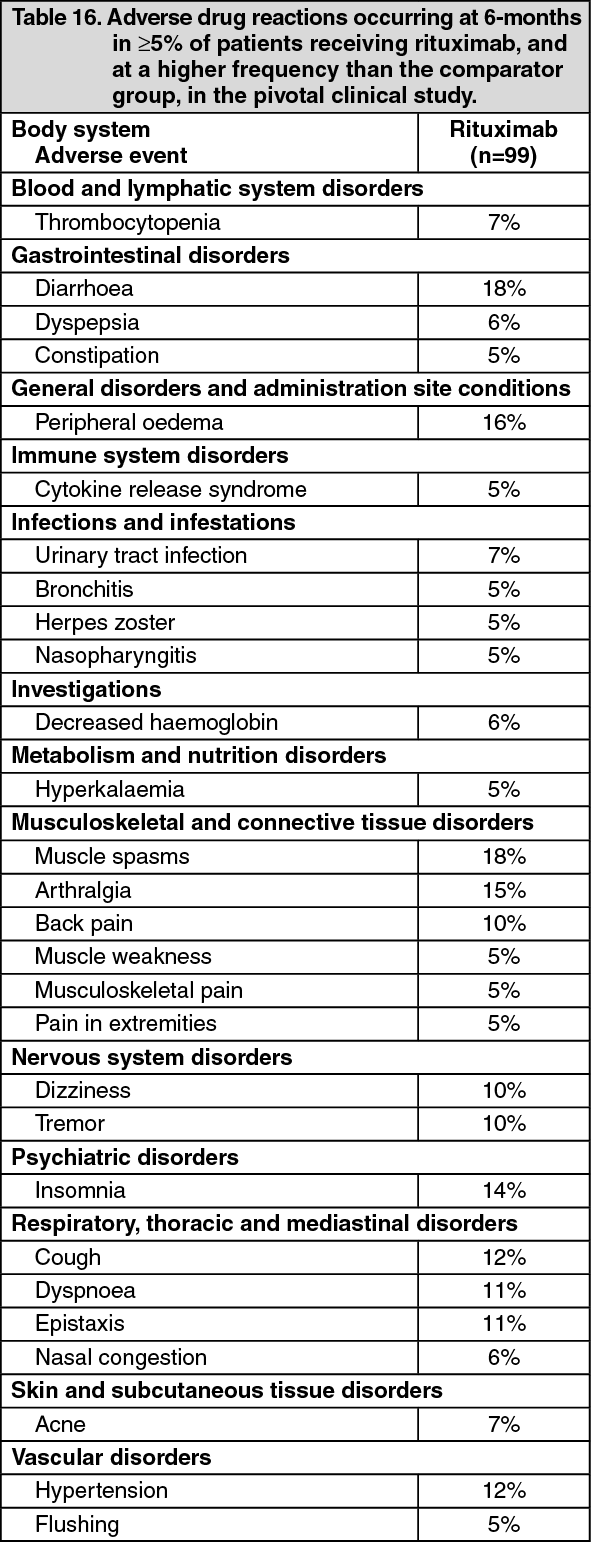 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageSelected adverse drug reactions: Infusion related reactions: IRRs in the GPA and MPA clinical trial were defined as any adverse event occurring within 24 hours of an infusion and considered to be infusion-related by investigators in the safety population. Ninety-nine patients were treated with rituximab and 12% experienced at least one IRR. All IRRs were CTC grade 1 or 2. The most common IRRs included cytokine release syndrome, flushing, throat irritation, and tremor. Rituximab was given in combination with intravenous glucocorticoids which may reduce the incidence and severity of these events.
Infections: In the 99 rituximab patients, the overall rate of infection was approximately 237 per 100 patient years (95% CI 197 - 285) at the 6-month primary endpoint. Infections were predominately mild to moderate and consisted mostly of upper respiratory tract infections, herpes zoster and urinary tract infections. The rate of serious infections was approximately 25 per 100 patient years. The most frequently reported serious infection in the rituximab group was pneumonia at a frequency of 4%.
Malignancies: The incidence of malignancy in rituximab treated patients in the granulomatosis with polyangiitis and microscopic polyangiitis clinical study was 2.00 per 100 patient years at the study common closing date (when the final patient had completed the follow-up period). On the basis of standardized incidence ratios, the incidence of malignancies appears to be similar to that previously reported in patients with ANCA-associated vasculitis.
Cardiovascular adverse reactions: Cardiac events occurred at a rate of approximately 273 per 100 patient years (95% CI 149-470) at the 6-month primary endpoint. The rate of serious cardiac events was 2.1 per 100 patient years (95% CI 3-15). The most frequently reported events were tachycardia (4%) and atrial fibrillation (3%) (see Precautions).
Neurologic events: Cases of posterior reversible encephalopathy syndrome (PRES) / reversible posterior leukoencephalopathy syndrome (RPLS) have been reported in autoimmune conditions. Signs and symptoms included visual disturbance, headache, seizures and altered mental status, with or without associated hypertension. A diagnosis of PRES/RPLS requires confirmation by brain imaging. The reported cases had recognised risk factors for PRES/RPLS, including the patients' underlying disease, hypertension, immunosuppressive therapy and/or chemotherapy.
Hepatitis-B reactivation: A small number of cases of hepatitis-B reactivation, some with fatal outcome, have been reported in granulomatosis with polyangiitis and microscopic polyangiitis patients receiving rituximab in the post-marketing setting.
Hypogammaglobulinaemia: Hypogammaglobulinaemia (IgA, IgG or IgM below the lower limit of normal) has been observed in granulomatosis with polyangiitis and microscopic polyangiitis patients treated with rituximab. At 6 months, in the active-controlled, randomised, double-blind, multicentre, non-inferiority trial, in the rituximab group, 27%, 58% and 51% of patients with normal immunoglobulin levels at baseline, had low IgA, IgG and IgM levels, respectively compared to 25%, 50% and 46% in the cyclophosphamide group. There was no increased rate in overall infections or serious infections in patients with low IgA, IgG or IgM.
Neutropenia: In the active-controlled, randomised, double-blind, multicentre, non-inferiority trial of rituximab in granulomatosis with polyangiitis and microscopic polyangiitis, 24% of patients in the rituximab group (single course) and 23% of patients in the cyclophosphamide group developed CTC grade 3 or greater neutropenia. Neutropenia was not associated with an observed increase in serious infection in rituximab-treated patients. The effect of multiple rituximab courses on the development of neutropenia in granulomatosis with polyangiitis and microscopic polyangiitis patients has not been studied in clinical trials.
Skin and subcutaneous tissue disorders: Toxic epidermal necrolysis (Lyell's syndrome) and Stevens-Johnson syndrome, some with fatal outcome, have been reported very rarely.
View ADR Monitoring Form