


 Sign Out
Sign Out

Pharmacology: Mechanism of action: Tasigna is a potent and selective inhibitor of the ABL tyrosine kinase activity of the BCR-ABL oncoprotein both in cell lines and in primary Philadelphia-chromosome positive leukemia cells. The drug binds strongly within the ATP-binding site in such a manner that it is a potent inhibitor of wild-type BCR-ABL and maintains activity against 32/33 imatinib-resistant mutant forms of BCR-ABL. As a consequence of this biochemical activity, nilotinib selectively inhibits the proliferation and induces apoptosis in BCR-ABL dependent cell lines and in primary Philadelphia-chromosome positive leukemia cells derived from CML patients. In murine models of CML, as a single agent nilotinib reduces tumor burden and prolongs survival following oral administration.
Pharmacodynamics: Tasigna has little or no effect against the majority of other protein kinases examined, including SRC, except for the platelet-derived growth factor (PDGF), KIT, colony stimulating factor 1 receptor (CSF1R), discoidin domain receptor (DDR) and Ephrin receptor kinases which it inhibits at concentrations within the range achieved following oral administration at therapeutic doses recommended for the treatment of CML (see Table 1).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageClinical Studies: Newly Diagnosed Ph+ CML-CP: An open label, multicenter, randomized Phase III study was conducted to determine the efficacy of Tasigna versus imatinib in adult patients with cytogenetically confirmed newly diagnosed Ph+ CML-CP. Patients were within six months of diagnosis and were previously untreated for CML-CP, except for hydroxyurea and/or anagrelide. In addition, patients were stratified according to Sokal risk score at time of diagnosis.
Efficacy was based on a total of 846 patients (283 patients in the imatinib 400 mg once daily group, 282 patients in the nilotinib 300 mg twice daily group, 281 patients in the nilotinib 400 mg twice daily group).
Baseline characteristics were well balanced between the 3 groups. Median age was 46 years in the imatinib group and 47 years in both nilotinib groups, with 12.4%, 12.8% and 10.0% were ≥65 years of age in imatinib, nilotinib 300 mg twice daily and nilotinib 400 mg twice daily treatment groups, respectively. There were slightly more male than female patients in all groups (55.8%, 56.0% and 62.3% in imatinib, nilotinib 300 mg twice daily and nilotinib 400 mg twice daily, respectively). More than 60% of all patients were Caucasian, and 25% were Asian.
The primary data analysis time point was when all 846 patients completed 12 months of treatment (or discontinued earlier). Subsequent analyses reflect when patients completed 24, 36, 48 and 60 months of treatment (or discontinued earlier). The median time on treatment was approximately 60 months in all three treatment groups. The median actual dose intensity was 400 mg/day in the imatinib group, 593 mg/day in the nilotinib 300 mg twice daily group and 773 mg/day in the nilotinib 400 mg twice daily group. This study is on-going.
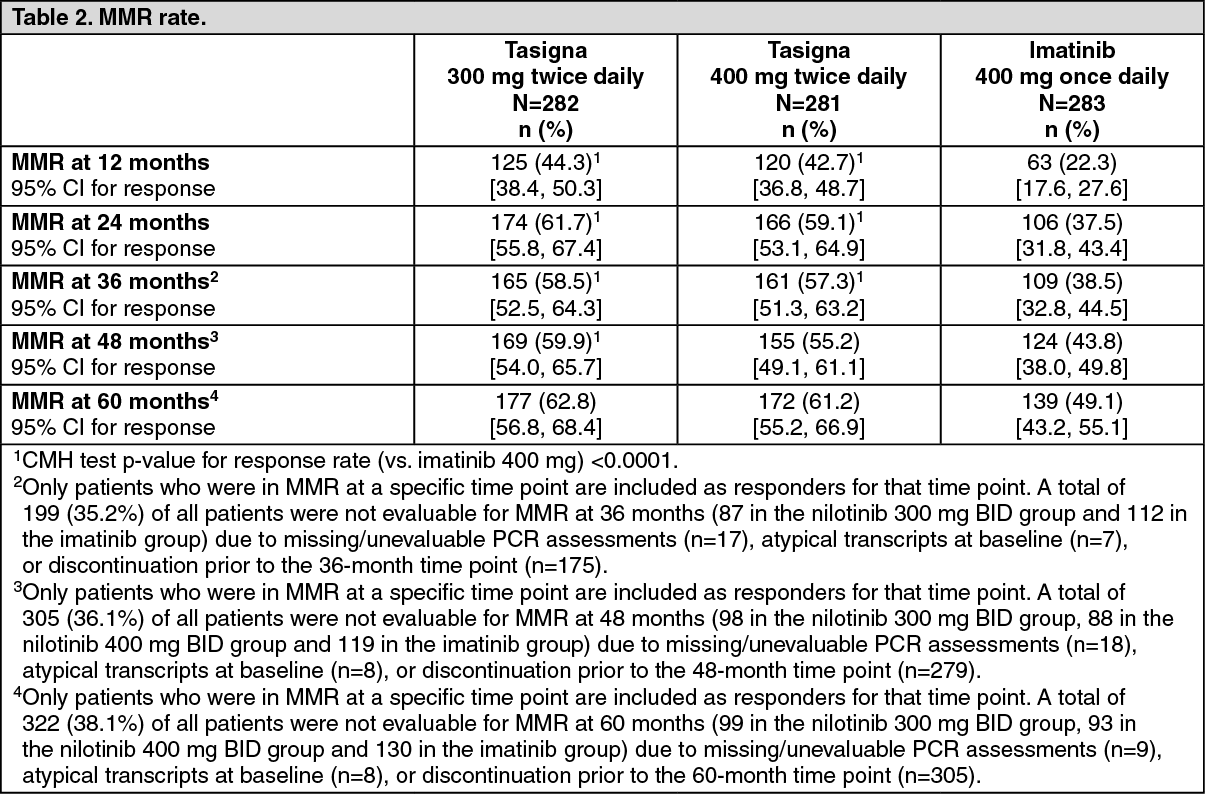
Major molecular response (MMR): The primary efficacy variable was MMR at 12 months after the start of study medication. MMR was defined as ≤0.1% BCR-ABL/ABL % by international scale measured by real-time quantitative polymerase chain reaction (RQ-PCR), which corresponds to a ≥3 log reduction of BCR-ABL transcript from standardized baseline.
The primary efficacy endpoint, MMR rate at 12 months was statistically significantly superior in the nilotinib 300 mg twice daily group compared to the imatinib 400 mg once daily group (44.3% vs. 22.3%, p<0.0001). The rate of MMR at 12 months, was also statistically significantly higher in the nilotinib 400 mg twice daily group compared to the imatinib 400 mg once daily group (42.7% vs. 22.3%, p<0.0001), Table 2.
At the nilotinib recommended dose of 300 mg twice daily, the rates of MMR at 3, 6, 9 and 12 months were 8.9%, 33.0%, 43.3% and 44.3%. In the nilotinib 400 mg twice daily group, the rates of MMR at 3, 6, 9 and 12 months were 5.0%, 29.5%, 38.1% and 42.7%. In the imatinib 400 mg once daily group, the rates of MMR at 3, 6, 9 and 12 months were 0.7%, 12.0%, 18.0% and 22.3%.
The MMR rates at 12, 24, 36, 48 and 60 months are presented in Table 2. (See Table 2.)
 Click on icon to see table/diagram/image
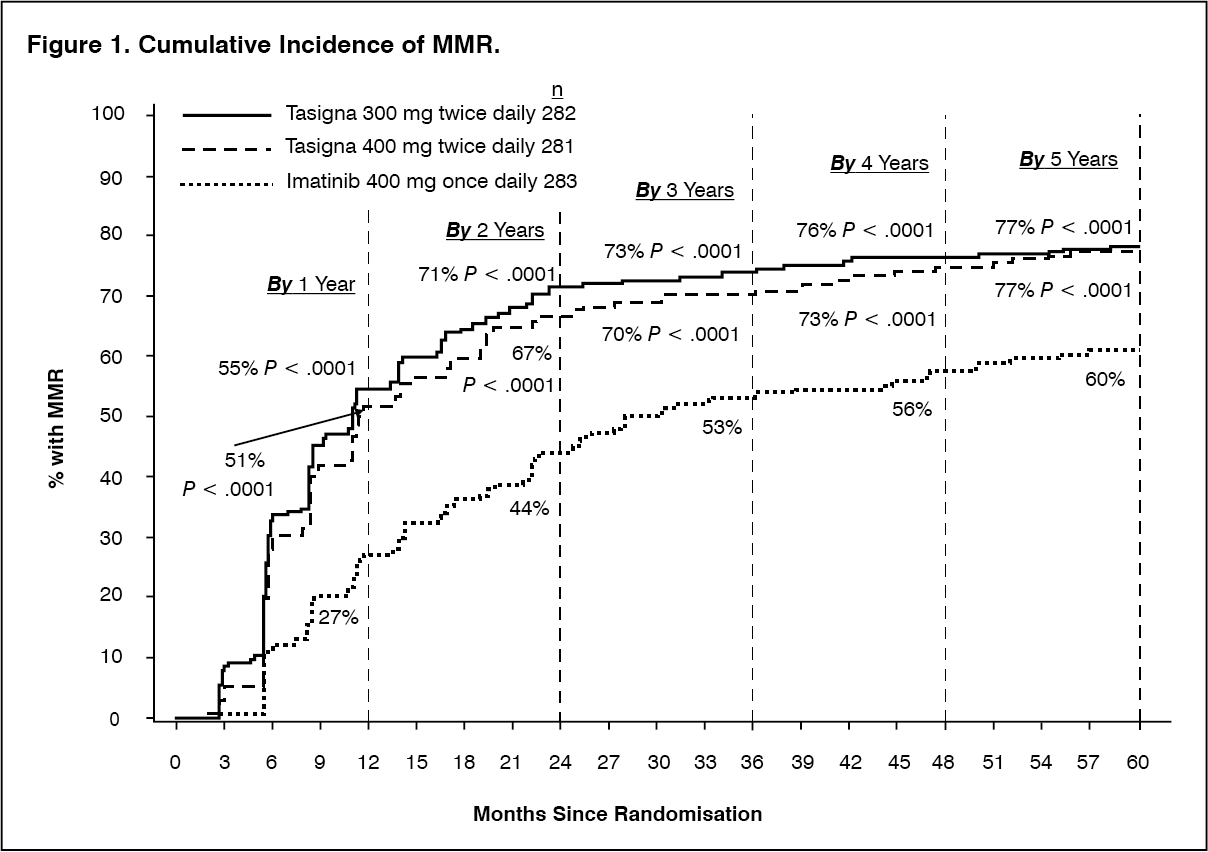
Click on icon to see table/diagram/imageMMR rates by different time points (including patients who achieved MMR at or before those time points as responders) are presented in the cumulative incidence of MMR (see Figure 1).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageFor all Sokal risk groups, the MMR rates at all time points remained consistently higher in the two nilotinib groups than in the imatinib group.
In a retrospective analysis, 91% (234/258) of patients on nilotinib 300 mg twice daily achieved BCR-ABL levels ≤10% at 3 months of treatment compared to 67% (176/264) of patients on imatinib 400 mg once daily. Patients with BCR-ABL levels ≤10% at 3 months of treatment show a greater overall survival at 60 months compared to those who did not achieve this molecular response level (97% vs. 82% respectively [p=0.0116]).
Based on the Kaplan-Meier analyses of time to first MMR among all patients the probability of achieving MMR at different time points were higher in both nilotinib groups compared to the imatinib group (hazard ratio/HR=2.20 and stratified log-rank p<0.0001 between nilotinib 300 mg twice daily and imatinib, HR=1.90 and stratified log-rank p<0.0001 between nilotinib 400 mg twice daily and imatinib).
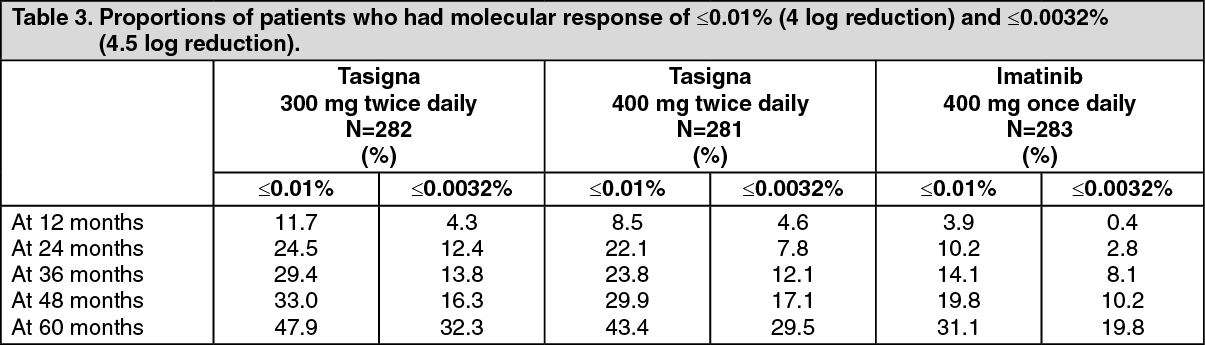
The proportions of patients who had a molecular response of ≤0.01% and ≤0.0032% by International Scale (IS) at different time-points is presented in Table 3 and the proportion of patients who had a molecular response of ≤0.01% and ≤0.0032% by IS-by different time-points are presented in Figures 2 and 3. Molecular responses of ≤0.01% and ≤0.0032% by IS corresponds to a ≥4 log reduction and ≥4.5 log reduction, respectively, of BCR-ABL transcripts from a standardized baseline. (See Table 3 and Figures 2 and 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image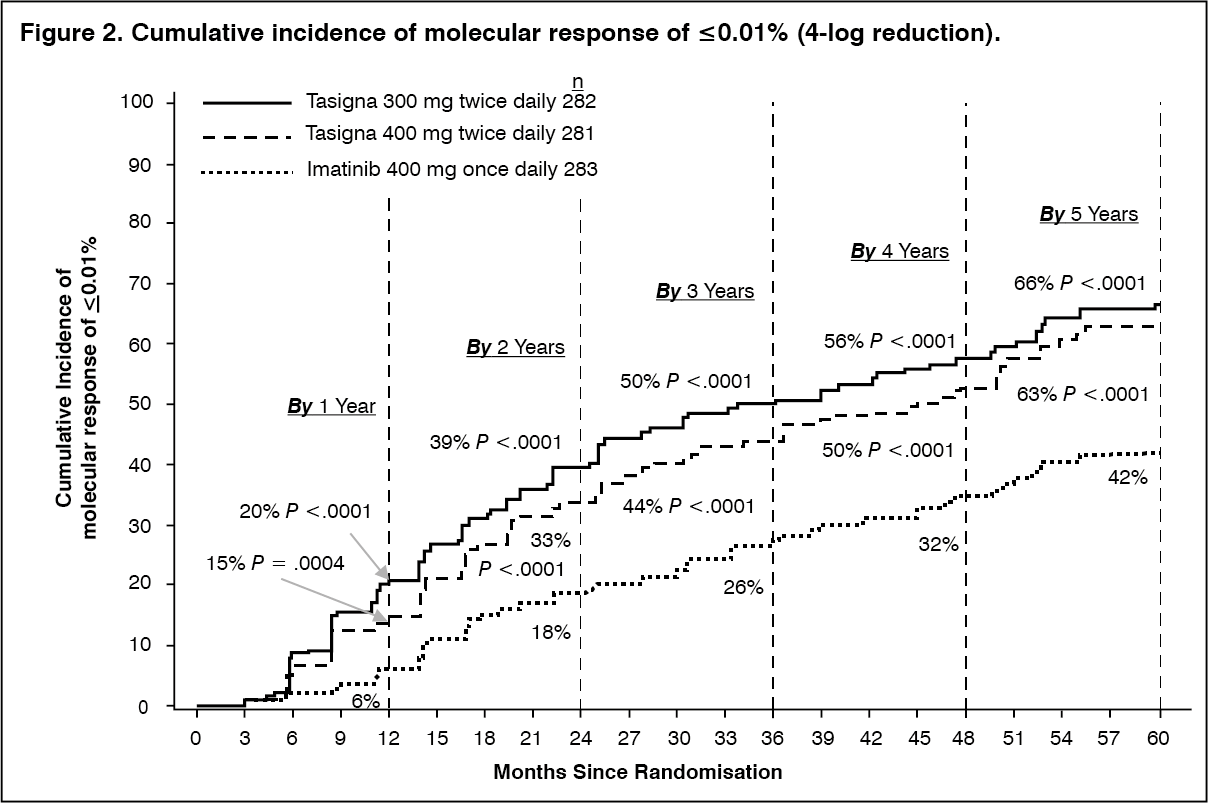 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image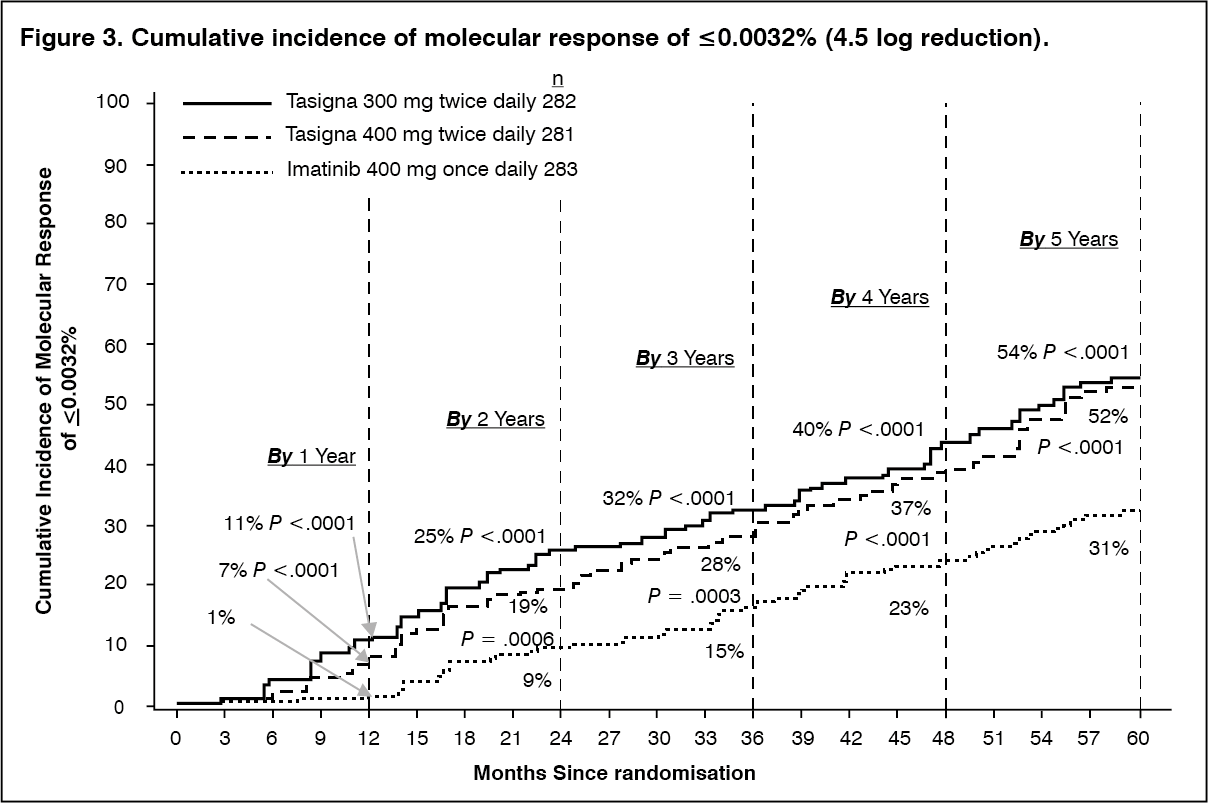 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageDuration of MMR: Based on Kaplan-Meier estimates of the duration of first MMR, the proportions of patients who were maintaining response after 60 months among patients who achieved MMR were 93.4% (95% CI: 89.9% to 96.9%) in the nilotinib 300 mg twice daily group, 92.0% (95% CI: 88.2% to 95.8%) in the nilotinib 400 mg twice daily group and 89.1% (95% CI: 84.2% to 94.0%) in the imatinib 400 mg once daily group.
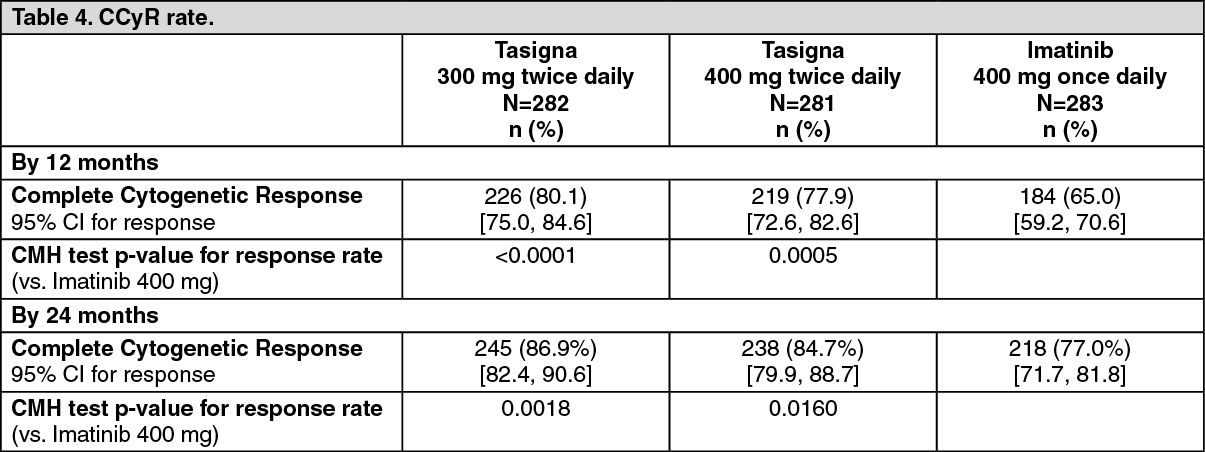
Complete cytogenetic response (CCyR): CCyR was defined as 0% Ph+ metaphases in the bone marrow based on a minimum of 20 metaphases evaluated. CCyR rate by 12 months (includes patients who achieved CCyR at or before the 12 month time point as responders) was statistically higher for both the nilotinib 300 mg twice daily and 400 mg twice daily groups compared to imatinib 400 mg once daily group, Table 4.
CCyR rate by 24 months (includes patients who achieved CCyR at or before the 24 month time point as responders) was statistically higher for both the nilotinib 300 mg twice daily and 400 mg twice daily groups compared to imatinib 400 mg once daily group. (See Table 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageDuration of CCyR: Based on Kaplan-Meier estimates, the proportions of patients who were maintaining response after 60 months among patients who achieved CCyR were 99.1% (95% CI: 97.9% to 100%) in the nilotinib 300 mg twice daily group, 98.7% (95% CI: 97.1% to 100%) in the nilotinib 400 mg twice daily group and 97.0% (95% CI: 94.7% to 99.4%) in the imatinib 400 mg once daily group.
Progression to AP/BC on Treatment: Progression to AP/BC on treatment is defined as the time from the date of randomization to the first documented disease progression to AP/BC or CML-related death. Overall by the cut-off date, 17 patients progressed to AP or BC on treatment (2 in the nilotinib 300 mg twice daily group, 3 in the nilotinib 400 mg twice daily group and 12 in the imatinib 400 mg once daily group). The estimated rates of patients free from progression to AP or BC at 60 months were 99.3%, 98.7% and 95.2%, respectively (HR=0.1599 and stratified log-rank p=0.0059 between nilotinib 300 mg twice daily (b.i.d) and imatinib, HR=0.2457 and stratified log-rank p=0.0185 between nilotinib 400 mg b.i.d and imatinib). No new events of progression to AP/BC were reported on-treatment since the 2-year analysis.
Including clonal evolution as a criterion for progression, a total of 25 patients progressed to AP or BC on treatment by the cut-off date (3 in the nilotinib 300 mg twice daily group, 5 in the nilotinib 400 mg twice daily group and 17 in the imatinib 400 mg once daily group). The estimated rates of patients free from progression to AP or BC including clonal evolution at 60 months were 98.7%, 97.9% and 93.2%, respectively (HR=0.1626 and stratified log-rank p=0.0009 between nilotinib 300 mg b.i.d and imatinib, HR=0.2848 and stratified log-rank p=0.0085 between nilotinib 400 mg b.i.d and imatinib).
Overall survival (OS): A total of 50 patients died during treatment or during the follow-up after discontinuation of treatment (18 in the nilotinib 300 mg twice daily group, 10 in the nilotinib 400 mg twice daily group and 22 in the imatinib 400 mg once daily group). Twenty-six (26) of these 50 deaths were related to CML (6 in the nilotinib 300 mg twice daily group, 4 in the nilotinib 400 mg twice daily group and 16 in the imatinib 400 mg once daily group). The estimated rates of patients alive at 60 months were 93.7%, 96.2% and 91.7%, respectively (HR=0.8026 and stratified log-rank p=0.4881 between nilotinib 300 mg twice daily and imatinib, HR=0.4395 and stratified log-rank p=0.0266 between nilotinib 400 mg twice daily and imatinib). Considering only CML-related deaths as events, the estimated rates of OS at 60 months were 97.7%, 98.5% and 93.8%, respectively (HR=0.3673 and stratified log-rank p=0.0292 between nilotinib 300 mg twice daily and imatinib, HR=0.2411 and stratified log-rank p=0.0057 between nilotinib 400 mg twice daily and imatinib).
Switch to Tasigna treatment in adult patients with Ph+ CML-CP who have not achieved a molecular response greater than or equal to a 4.5-log reduction with imatinib treatment: In an open-label, multicenter, randomized Phase III study, 207 adult patients with Ph+ CML-CP who received treatment with imatinib for at least 2 years, with no permanent imatinib dose adjustment within 6 months and no major toxicity within 3 months of study entry were enrolled in the study. Patients were randomized 1:1 either to receive Tasigna 400 mg twice daily (n=104) or to continue treatment with imatinib at the same dose (400 mg or 600 mg once daily) as administered prior to randomization (n=103). Randomization was stratified by duration of prior treatment with imatinib and duration of prior interferon use. The median time on treatment (from first day of treatment to last day of randomized treatment) at cut-off was 47.2 months in the Tasigna treatment arm, and 37.0 months and 26.7 months in the 400 mg and 600 mg dose cohorts of the imatinib arm, respectively.
The two treatment arms were well balanced with respect to demographic and baseline characteristics (including BCR-ABL transcript levels at study entry). Median age was 46 years in the Tasigna arm and 52 years in the imatinib arm, with 13.5% and 13.6% of patients aged ≥65 years in the Tasigna and imatinib treatment arms, respectively. There were more male (68.3% in the Tasigna treatment arm and 63.1% in the imatinib treatment arm) than female patients. More than 80% of all patients were Caucasians. Up to the cut-off date, the median actual dose intensity was 775.7 mg/day in the Tasigna treatment arm and 400 mg/day and 600 mg/day in the two dose cohorts of the imatinib treatment arm, respectively.
The primary endpoint of the study was the rate of confirmed best cumulative complete molecular response (CMR) within the first year of study therapy with Tasigna or imatinib. The rate of confirmed best cumulative CMR during the first 12 months was 12.5% in the Tasigna arm and 5.8% in the imatinib arm. The primary endpoint did not reach statistical significance at the early 12-month time point (p=0.1083), with an odds ratio (OR) of 2.096 in favor of Tasigna.
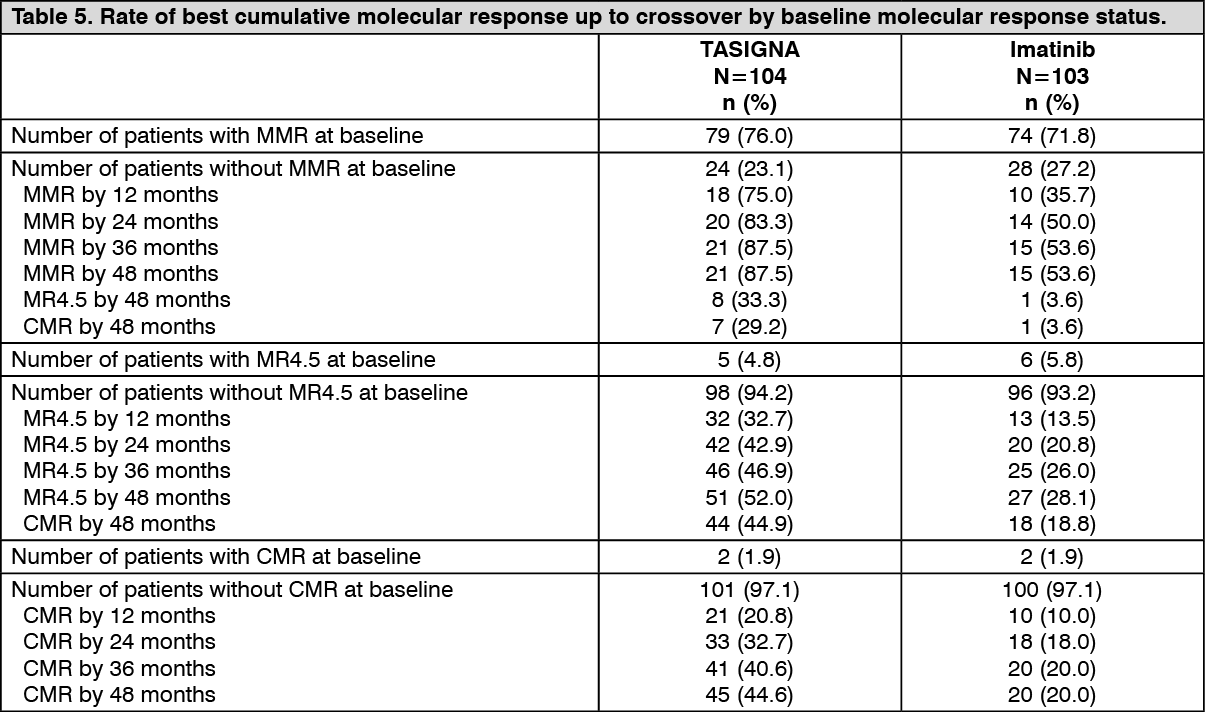
Longer-term follow-up of the primary outcome variable at 48-months was a secondary endpoint. Analyses conducted to assess the achievement of different levels of molecular response up to crossover in patients without the corresponding response at baseline showed that switching from imatinib to Tasigna was associated with a clinically meaningful increase in the numbers of patients attaining MMR, MR4.5, and CMR under their randomized treatment at Month 48 (see Table 5).
 Click on icon to see table/diagram/image
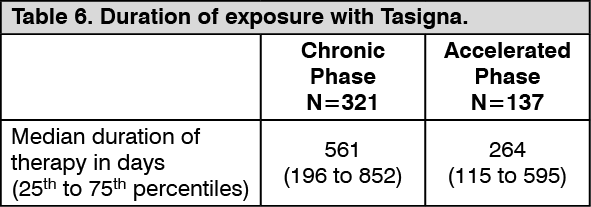
Click on icon to see table/diagram/imageResistance or intolerant Ph+ CML: An open-label multicenter Phase II study was conducted to determine the efficacy of Tasigna (400 mg twice daily) in adult patients with imatinib resistant or intolerant CML with separate treatment arms for chronic and accelerated phase disease. Efficacy was based on 321 CP patients and 137 AP patients enrolled. Median duration of treatment was 561 days and 264 days, respectively (see Table 6). Tasigna was administered on a continuous basis, (twice daily 2 hours after a meal and no additional food for at least one hour) unless there was evidence of inadequate response or disease progression. Dose escalation to 600 mg twice daily was allowed. (See Table 6.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageResistance to imatinib included failure to achieve a complete hematologic response (CHR) (by 3 months), cytogenetic response (by 6 months) or major cytogenetic response (by 12 months) or progression of disease after a previous cytogenetic or hematologic response. Imatinib intolerance included patients who discontinued imatinib because of toxicity and were not in major cytogenetic response at time of study entry.
Overall, 73% of patients were imatinib-resistant while 27% were imatinib-intolerant. The majority of patients had a long history of CML that included extensive prior treatment with other antineoplastic agents such as imatinib, hydroxyurea, interferon, and some that had even failed stem cell transplant (Table 7). The median highest prior imatinib dose had been 600 mg/day for CP and AP patients, and the highest prior imatinib dose was ≥600 mg/day in 74% of all patients with 40% of patients receiving imatinib doses ≥800 mg/day. (See Table 7.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe primary endpoint in the CP patients was major cytogenetic response (MCyR), defined as elimination (CCyR, complete cytogenetic response) or significant reduction to <35% Ph+ metaphases (partial cytogenetic response) of Ph+ hematopoietic cells. Complete hematologic response in CP patients was evaluated as a secondary endpoint. The primary endpoint in the AP patients was overall confirmed hematologic response, defined as either a complete hematologic response, no evidence of leukemia or return to chronic phase.
Chronic Phase: The MCyR rate in 321 CP patients was 59%. Most responders achieved their MCyR rapidly within 3 months (median 2.8 months) of starting Tasigna treatment and these were sustained. The CCyR rate was 44%. The median time to achieve CCyR was just past 3 months (median 3.3 months). Of the patients who achieved MCyR, 77% (95% CI: 71% to 84%) were maintaining response at 24 months. Median duration of MCyR has not been reached. Of the patients who achieved CCyR, 84% (95% CI: 77% to 91%) were maintaining response at 24 months. Median duration of CCyR has not been reached. Patients with a CHR at baseline achieved a MCyR faster (1.4 vs. 2.8 months). Of CP patients without a baseline CHR, 76% achieved a CHR, median time to CHR was 1 month and median duration of CHR has not been reached.
The estimated 24-month overall survival rate in CML-CP patients was 87%.
Accelerated Phase: The overall confirmed HR rate in 137 AP patients was 55%. Most responders achieved a HR early with Tasigna treatment (median 1.0 months) and these have been durable (median duration of confirmed HR was 21.5 months). Of the patients who achieved HR, 49% (95% CI: 35% to 62%) were maintaining response at 24 months. MCyR rate was 32% with a median time to response of 2.8 months. Of the patients who achieved MCyR, 66% (95% CI: 50% to 82%) were maintaining response at 24 months. Median duration of MCyR has not been reached. The rates of response for the two treatment arms are reported in Table 8.
The estimated 24-month overall survival rate in CML-AP patients was 70%. (See Table 8.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageSeparate treatment arms were also included in the Phase II study to study Tasigna in a group of CP and AP patients who had been extensively pre-treated with multiple therapies including a tyrosine kinase inhibitor agent in addition to imatinib. Of these patients 30/36 (83%) were treatment-resistant. In 22 CP patients evaluated for efficacy Tasigna induced a 32% MCyR rate and a 50% CHR rate. In 11 AP patients evaluated for efficacy, treatment induced a 36% overall HR rate.
After imatinib failure, 24 different BCR-ABL mutations were noted in 42% of chronic phase and 54% of accelerated phase CML patients who were evaluated for mutations. Tasigna demonstrated efficacy in patients harboring a variety of BCR-ABL mutations associated with imatinib resistance, except T315I.
Treatment discontinuation in newly diagnosed Ph+ CML-CP adult patients who have achieved a sustained deep molecular response: In an open-label, multicenter, single-arm study, 215 adult patients with Ph+ CML-CP treated with Tasigna in first-line for ≥2 years who achieved MR4.5 as measured with the MolecularMD MRDx BCR-ABL Test were enrolled to continue Tasigna treatment for an additional 52 weeks (Tasigna consolidation phase). Of the 215 patients, 190 patients (88.4%) entered the TFR phase after achieving a sustained deep molecular response during the consolidation phase, defined by the following criteria: The 4 last quarterly assessments (taken every 12 weeks) were at least MR4.0 (BCR-ABL/ABL ≤0.01% IS), and maintained for 1 year; The last assessment being MR4.5 (BCR-ABL/ABL ≤0.0032% IS); No more than two assessments falling between MR4.0 and MR4.5 (0.0032% IS <BCR-ABL/ABL ≤0.01% IS).
In the set of patients who entered the TFR phase, the median age was 55 years. The proportion of female patients was 49.5%, and 21.1% of the patients were ≥65 years of age. The median actual dose intensity during the 52-week Tasigna consolidation phase was 600.0 mg/day.
BCR-ABL levels were monitored every 4 weeks during the first 48 weeks of the TFR phase. Monitoring frequency was intensified to every 2 weeks upon the loss of MR4.0. Biweekly monitoring ended at one of the following time points: Loss of MMR requiring patient to re-initiate Tasigna treatment; When the BCR-ABL levels returned to a range between MR4.0 and MR4.5; When the BCR-ABL levels remained lower than MMR for 4 consecutive measurements (8 weeks from initial loss of MR4.0).
Any patient with loss of MMR during the TFR phase re-initiated Tasigna treatment at 300 mg twice daily or at a reduced dose level of 400 mg once daily if required from the perspective of tolerance, within 5 weeks after the collection date of the blood sample demonstrating loss of MMR. Patients who required re-initiation of Tasigna treatment were monitored for BCR-ABL levels every 4 weeks for the first 24 weeks and then every 12 weeks thereafter in patients who regained MMR.
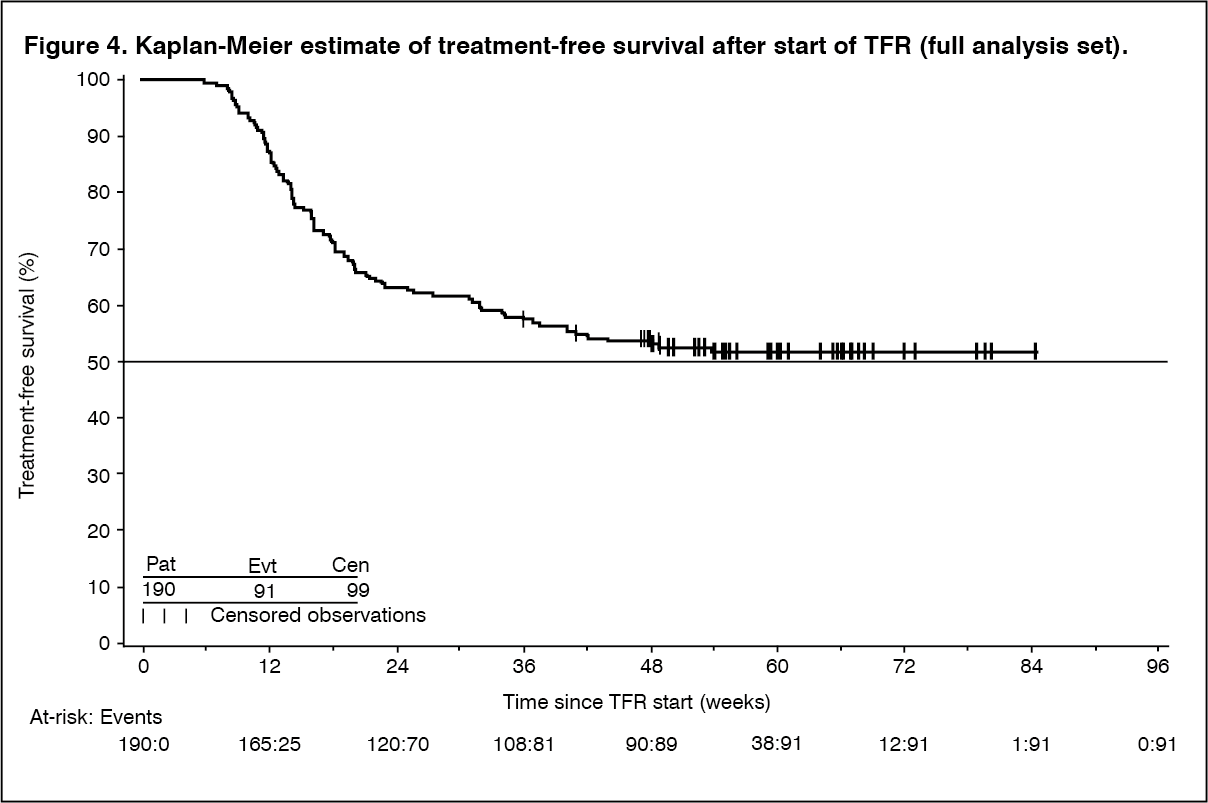
The primary endpoint was the percentage of patients who were in MMR at 48 weeks after starting the TFR phase (considering any patient who required re-initiation of treatment as non-responder). Of the 190 patients who entered the TFR phase, 98 patients (51.6% [95% CI: 44.2, 58.9]) were in MMR in the TFR phase at 48 weeks.
Eighty-eight patients (46.3%) discontinued from the TFR phase due to loss of MMR, and 1 (0.5%), 1 (0.5%), and 3 patients (1.6%) due to death from unknown cause, physician decision, and subject decision, respectively. Among the 88 patients who discontinued the TFR phase due to loss of MMR, 86 patients restarted Tasigna treatment and 2 patients permanently discontinued from the study.
Of the 86 patients who restarted treatment due to loss of MMR in the TFR phase, 85 patients (98.8%) regained MMR, (one patient discontinued study permanently due to subject decision) and 76 patients (88.4%) regained MR4.5 by the time of the cut-off date.
The Kaplan-Meier (KM) estimated median time on Tasigna to regain MMR and MR4.5 was 7.9 weeks (95% CI: 5.1, 8.0) and 13.1 weeks (95% CI: 12.3, 15.7), respectively. The KM estimated MMR rate at 24 weeks of re-initiation was 98.8% (95% CI: 94.2, 99.9). The KM estimated MR4.5 rate at 24 weeks of re-initiation was 90.9% (95% CI: 83.2, 96.0).
Among the 190 patients in the TFR phase, 99 patients (52.1%) did not have a treatment-free survival (TFS) event on or before the 48 month cut-off date, and were censored at the date of their last assessment prior to cut-off. The KM estimate of median TFS has not yet been reached (Figure 4). (See Figure 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageTreatment discontinuation in Ph+ CML-CP adult patients who have achieved a sustained deep molecular response on Tasigna following prior imatinib therapy: In an open-label, multicenter, single-arm study, 163 adult patients with Ph+ CML-CP taking TKIs for ≥3 years (imatinib as initial TKI therapy for more than 4 weeks without documented MR4.5 on imatinib at the time of switch to Tasigna, then switched to Tasigna for at least two years), and who achieved MR4.5 on Tasigna treatment as measured with the MolecularMD MRDx BCR-ABL Test were enrolled to continue Tasigna treatment for an additional 52 weeks (Tasigna consolidation phase). Of the 163 patients, 126 patients (77.3%) entered the TFR phase after achieving a sustained deep molecular response during the consolidation phase, defined by the following criterion: The 4 last quarterly assessments (taken every 12 weeks) showed no confirmed loss of MR4.5 (BCR-ABL/ABL ≤0.0032% IS) during 1 year.
The median age of the patients who entered the TFR phase was 56 years. The proportion of female patients was 55.6%, and 27.8% of the patients were ≥65 years of age. The median actual dose intensity during the 52-week Tasigna consolidation phase was 771.8 mg/day with 52.4% and 29.4% of patients receiving a daily Tasigna dose of 800 mg and 600 mg just before entry into the TFR phase, respectively.
Patients who entered the TFR phase but experienced two consecutive measurements of BCR-ABL/ABL >0.01% IS were considered having a confirmed loss of MR4.0, triggering re-initiation of Tasigna treatment. Patients with loss of MMR in the TFR phase immediately restarted Tasigna treatment without confirmation. All patients who restarted Tasigna therapy had BCR-ABL transcript levels monitored every 4 weeks for the first 24 weeks, then once every 12 weeks.
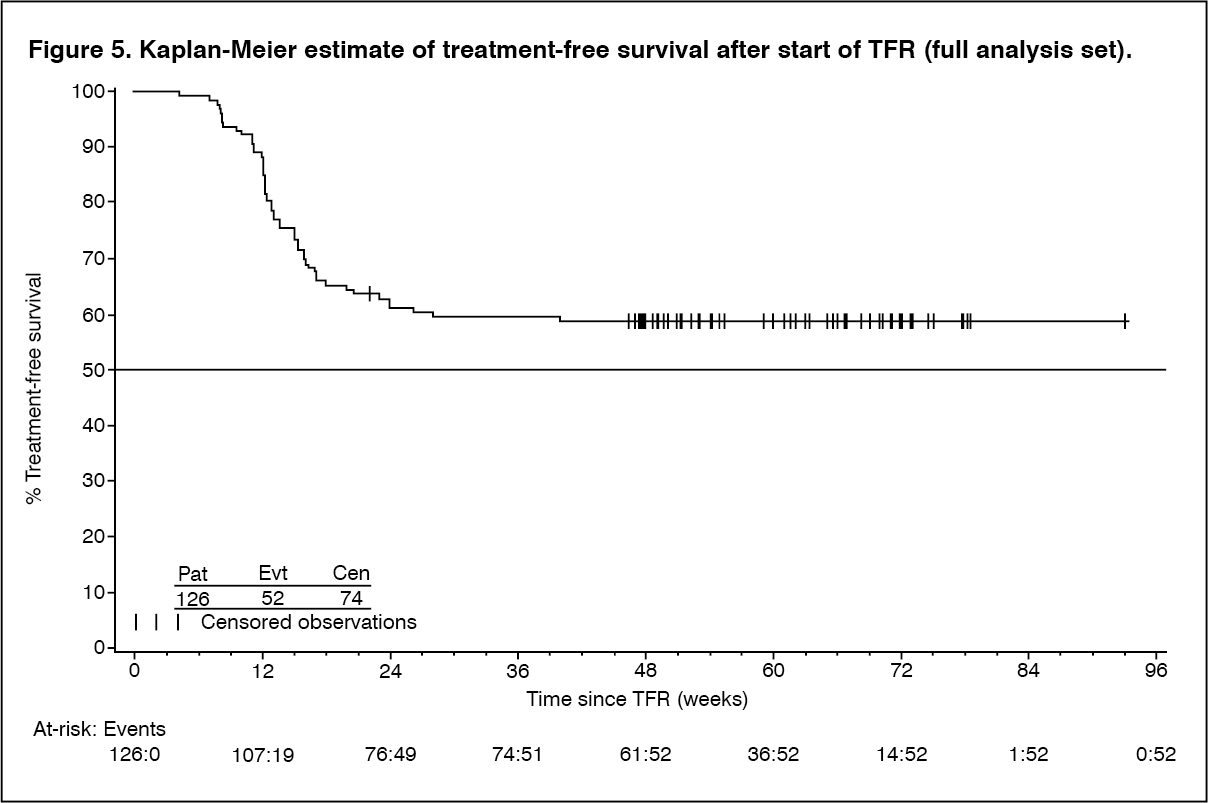
The primary endpoint was defined as the proportion of patients without confirmed loss of MR4.0 or loss of MMR within 48 weeks following discontinuation of Tasigna therapy. Of the 126 patients who entered the TFR phase, 73 patients (57.9%, [95% CI: 48.8, 66.7]) had no loss of MMR, no confirmed loss of MR4.0, and no re-initiation of Tasigna therapy within 48 weeks after the start of the TFR phase.
Among the 53 patients who discontinued from the TFR phase due to confirmed loss of MR4.0 or loss of MMR, 51 patients restarted Tasigna therapy and 2 patients permanently discontinued from the study. Of the 51 patients who restarted Tasigna treatment due to confirmed loss of MR4.0 or loss of MMR in the TFR phase, 48 patients (94.1%) regained MR4.0 and 3 patients (5.9%) did not regain MR4.0. Forty-seven patients (92.2%) regained MR4.5 and 4 patients (7.8%) did not regain MR4.5 by the time of the cut-off date.
The Kaplan-Meier (KM) estimated median time on Tasigna to regain MR4.0 and MR4.5 was 12.0 weeks (95% CI: 8.3, 12.7) and 13.1 weeks (95% CI: 12.4, 16.1), respectively. The KM estimated rate of MR4.0 at 48 weeks of re-initiation was 100.0%. (95% CI: not estimated). The KM estimated rate of MR4.5 at 48 weeks of re-initiation was 94.8% (95% CI: 85.1, 99.0).
Among the 126 patients in the TFR phase, 74 patients (58.7%) did not have a treatment-free survival (TFS) event on or before the 48-month cut-off date, and were censored at the date of their last assessment prior to cut-off. The other 52 patients had a TFS event (18 patients had confirmed loss of MR4.0, and 34 patients lost MMR). The median TFS has not yet been reached (Figure 5). (See Figure 5.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePharmacokinetics: Absorption: Peak concentrations of nilotinib are reached 3 hours after oral administration. Nilotinib absorption following oral administration was approximately 30%. The absolute bioavailability of nilotinib has not been determined. As compared to an oral drink solution (pH of 1.2 to 1.3), relative bioavailability of nilotinib capsule is approximately 50%. In healthy volunteers, Cmax and area under the concentration-time curve (AUC) of nilotinib are increased by 112% and 82%, respectively compared to fasting conditions when Tasigna is given with food. Administration of Tasigna 30 minutes or 2 hours after food increased bioavailability of nilotinib by 29% or 15%, respectively (see Dosage & Administration, Precautions and Interactions). Nilotinib absorption (relative bioavailability) may be reduced by approximately 48% and 22% in patients with total gastrectomy and partial gastrectomy, respectively.
Distribution: Blood-to-plasma ratio of nilotininb is 0.68. Plasma protein-binding is approximately 98% on the basis of in vitro experiments.
Biotransformation/metabolism: Main metabolic pathways identified in healthy subjects are oxidation and hydroxylation. Nilotinib is the main circulating component in the serum. None of the metabolites contribute significantly to the pharmacological activity of nilotinib.
Elimination: After a single dose of radiolabelled nilotinib in healthy subjects, greater than 90% of the dose was eliminated within 7 days mainly in feces. Parent drug accounted for 69% of the dose.
The apparent elimination half-life estimated from the multiple dose PK with daily dosing was approximately 17 hours. Inter-patient variability in nilotinib PK was moderate to high (%CV: 33% to 43%).
Linearity/non-linearity: Steady-state nilotinib exposure was dose-dependent with less than dose-proportional increases in systemic exposure at dose levels higher than 400 mg given as once daily dosing. Daily systemic exposure to nilotinib of 400 mg twice-daily dosing at steady-state was 35% higher than with 800 mg once-daily dosing. Systemic exposure (AUC) of nilotinib at steady-state at a dose level of 400 mg twice daily was approximately 13.4% higher than with 300 mg twice daily. The average nilotinib trough and peak concentrations over 12 months were approximately 15.7% and 14.8% higher following 400 mg twice daily dosing compared to 300 mg twice daily. There was no relevant increase in exposure to nilotinib when the dose was increased from 400 mg twice-daily to 600 mg twice-daily.
Steady state conditions were essentially achieved by day 8. An increase in systemic exposure to nilotinib between the first dose and steady-state was approximately 2-fold for the 400 mg once daily dosing and 3.8-fold for the 400 mg twice-daily dosing.
Bioavailability/bioequivalence studies: Single-dose administration of 400 mg of nilotinib, using 2 capsules of 200 mg whereby the content of each capsule was dispersed in one teaspoon of applesauce, was shown to be bioequivalent with a single dose administration of 2 intact capsules of 200 mg.
Toxicology: Non-Clinical Safety Data: Nilotinib has been evaluated in safety pharmacology, repeated dose toxicity, genotoxicity, reproductive toxicity (see Use in Pregnancy & Lactation), phototoxicity and carcinogenicity (rat and mice) studies.
Safety pharmacology and repeated dose toxicity: Nilotinib did not have effects on central nervous system (CNS) or respiratory functions. In vitro cardiac safety studies demonstrated a preclinical signal for QT prolongation. No effects were seen in ECG measurements in dogs or monkeys treated up to 39 weeks or in a special telemetry study in dogs.
Repeated dose toxicity studies in dogs up to 4 weeks duration and in cynomolgus monkeys up to 9 months duration, revealed the liver as the primary target organ of toxicity of nilotinib. Alterations included increased alanine aminotransferase and alkaline phosphatase activity, and histopathology findings (mainly sinusoidal cell or Kupffer cell hyperplasia/hypertrophy, bile duct hyperplasia and periportal fibrosis). In general the changes in clinical chemistry were fully reversible after a four week recovery period, the histological alterations only showed partial reversibility. Exposures at the lowest dose levels where the liver effects were seen were lower than the exposure in humans at a dose of 800 mg/day. Only minor liver alterations were seen in mice or rats treated up to 26 weeks. Mainly reversible increases in cholesterol levels were seen in rats, dogs and monkeys.
Carcinogenicity and mutagenicity: Genotoxicity studies in bacterial in vitro systems and in mammalian in vitro and in vivo systems with and without metabolic activation did not reveal any evidence for a mutagenic potential of nilotinib.
In the 2-year rat carcinogenicity study there was no evidence of carcinogenicity upon administration of nilotinib at 5, 15 and 40 mg/kg/day. Exposures (in terms of AUC) at the highest dose level were representing approximately 2 to 3 times human daily steady-state exposure (based on AUC) to nilotinib at the dose of 800 mg/day. The major target organ for non-neoplastic lesions was the uterus (dilatation, vascular ectasia, hyperplasia endothelial cell, inflammation and/or epithelial hyperplasia).
In the 26-week Tg.rasH2 mouse carcinogenicity study, in which nilotinib was administered at 30, 100 and 300 mg/kg/day, skin papillomas/carcinomas were detected at 300 mg/kg, representing approximately 30 to 40 times (based on AUC) the human exposure at the maximum approved dose of 800 mg/day (administered as 400 mg twice daily). The No-Observed-Effect-Level (NOEL) for the skin neoplastic lesions was 100 mg/kg/day, representing approximately 10 to 20 times the human exposure at the maximum approved dose of 800 mg/day (administered as 400 mg twice daily). The major target organs for non-neoplastic lesions were the skin (epidermal hyperplasia), the growing teeth (degeneration/atrophy of the enamel organ of upper incisors and inflammation of the gingiva/odontogenic epithelium of incisors) and the thymus (increased incidence and/or severity of decreased lymphocytes).
Juvenile animal studies: In a juvenile development study, nilotinib was administered via oral gavage to juvenile rats from the first week postpartum through young adult (day 70 postpartum) at doses of 2, 6 and 20 mg/kg/day. Effects were limited to the dose of 20 mg/kg/day and consisted of reductions in body weight parameters and food consumption with recovery after dosing ceased. The NOEL in juvenile rats was considered to be 6 mg/kg/day. Overall, the toxicity profile in juvenile rats was comparable to that observed in adult rats.
Phototoxicity: Nilotinib was shown to absorb light in the UV-B and UV-A range, and to be distributed into the skin showing a phototoxic potential in vitro. However, no phototoxicity has been observed in vivo. Therefore the risk that nilotinib causes photosensitization in patients is considered very low.