


 Sign Out
Sign Out
PHARMACOLOGY: Pharmacodynamics: Mechanism of Action: Binding of PD-L1 to the PD-1 and B7.1 receptors found on T cells suppresses cytotoxic T-cell activity through the inhibition of T-cell proliferation and cytokine production. PD-L1 may be expressed on tumor cells and tumor-infiltrating immune cells, and can contribute to the inhibition of the anti-tumor immune response in the microenvironment.
Atezolizumab is an Fc-engineered humanized immunoglobulin G1 (IgG1) monoclonal antibody that directly binds to PD-L1 and blocks interactions with the PD-1 and B7.1 receptors, releasing PD-L1 / PD-1 pathway-mediated inhibition of the immune response, including reactivating the anti-tumor immune response. Atezolizumab leaves the PD-L2 / PD-1 interaction intact. In syngeneic mouse tumor models, blocking PD-L1 activity resulted in decreased tumor growth.
Clinical/Efficacy Studies: UC: IMvigor 211: A phase III, open-label, multi-center, international, randomized study, GO29294 (IMvigor211), was conducted to evaluate the efficacy and safety of Tecentriq compared with chemotherapy (investigator's choice of vinflunine, docetaxel, or paclitaxel) in patients with locally advanced or metastatic urothelial carcinoma who progressed during or following a platinum-containing regimen. This study excluded patients who had a history of autoimmune disease; active or corticosteroid-dependent brain metastases; administration of a live, attenuated vaccine within 28 days prior to enrolment; and administration of systemic immunostimulatory agents within 4 weeks or systemic immunosuppressive medicinal product within 2 weeks prior to enrolment. Tumor assessments were conducted every 9 weeks for the first 54 weeks, and every 12 weeks thereafter. Tumor specimens were evaluated prospectively for PD-L1 expression on tumour-infiltrating immune cells (IC) and the results were used to define the PD-L1 expression subgroups for the analyses described as follows.
A total of 931 patients were enrolled. Patients were randomized (1:1) to receive either Tecentriq or chemotherapy. Randomization was stratified by chemotherapy (vinflunine vs taxane), PD-L1 expression status on IC (< 5% vs ≥ 5%), number of prognostic risk factors (0 vs. 1-3), and liver metastases (yes vs. no). Prognostic risk factors included time from prior chemotherapy of < 3 months, ECOG performance status > 0 and hemoglobin < 10 g/dL.
Tecentriq was administered as a fixed dose of 1200 mg by intravenous infusion every 3 weeks. No dose reduction of Tecentriq was allowed. Patients were treated until loss of clinical benefit as assessed by the investigator or unacceptable toxicity. Vinflunine was administered 320 mg/m2 by intravenous infusion on day 1 of each 3-week cycle until disease progression or unacceptable toxicity. Paclitaxel was administered 175 mg/m2 by intravenous infusion over 3 hours on day 1 of each 3-week cycle until disease progression or unacceptable toxicity. Docetaxel was administered 75 mg/m2 by intravenous infusion on day 1 of each 3-week cycle until disease progression or unacceptable toxicity. For all treated patients, the median duration of treatment was 2.8 months for the Tecentriq arm, 2.1 months for the vinflunine and paclitaxel arms and 1.6 months for the docetaxel arm.
The demographic and baseline disease characteristics of the primary analysis population were well balanced between the treatment arms. The median age was 67 years (range: 31 to 88), and 77.1% of patients were male. The majority of patients were white (72.1%), 53.9% of patients within the chemotherapy arm received vinflunine, 71.4% of patients had at least one poor prognostic risk factor and 28.8% had liver metastases at baseline. Baseline ECOG performance status was 0 (45.6%) or 1 (54.4%). Bladder was the primary tumor site for 71.1% of patients and 25.4% of patients had upper tract urothelial carcinoma. There were 24.2% of patients who received only prior platinum-containing adjuvant or neoadjuvant therapy and progressed within 12 months.
The primary efficacy endpoint for IMvigor211 was overall survival (OS). Secondary efficacy endpoints were objective response rate (ORR), progression-free survival (PFS), and duration of response (DOR). OS comparisons between the treatment arm and control arm were tested using a hierarchical fixed-sequence procedure based on a stratified log-rank test at two-sided level of 5% as follows: step 1) PD-L1 expression ≥5% subgroup, step 2) PD-L1 expression ≥1% subgroup, step 3) all comers. OS results for each of steps 2 and 3 could only be formally tested if the result in the preceding step was statistically significant.
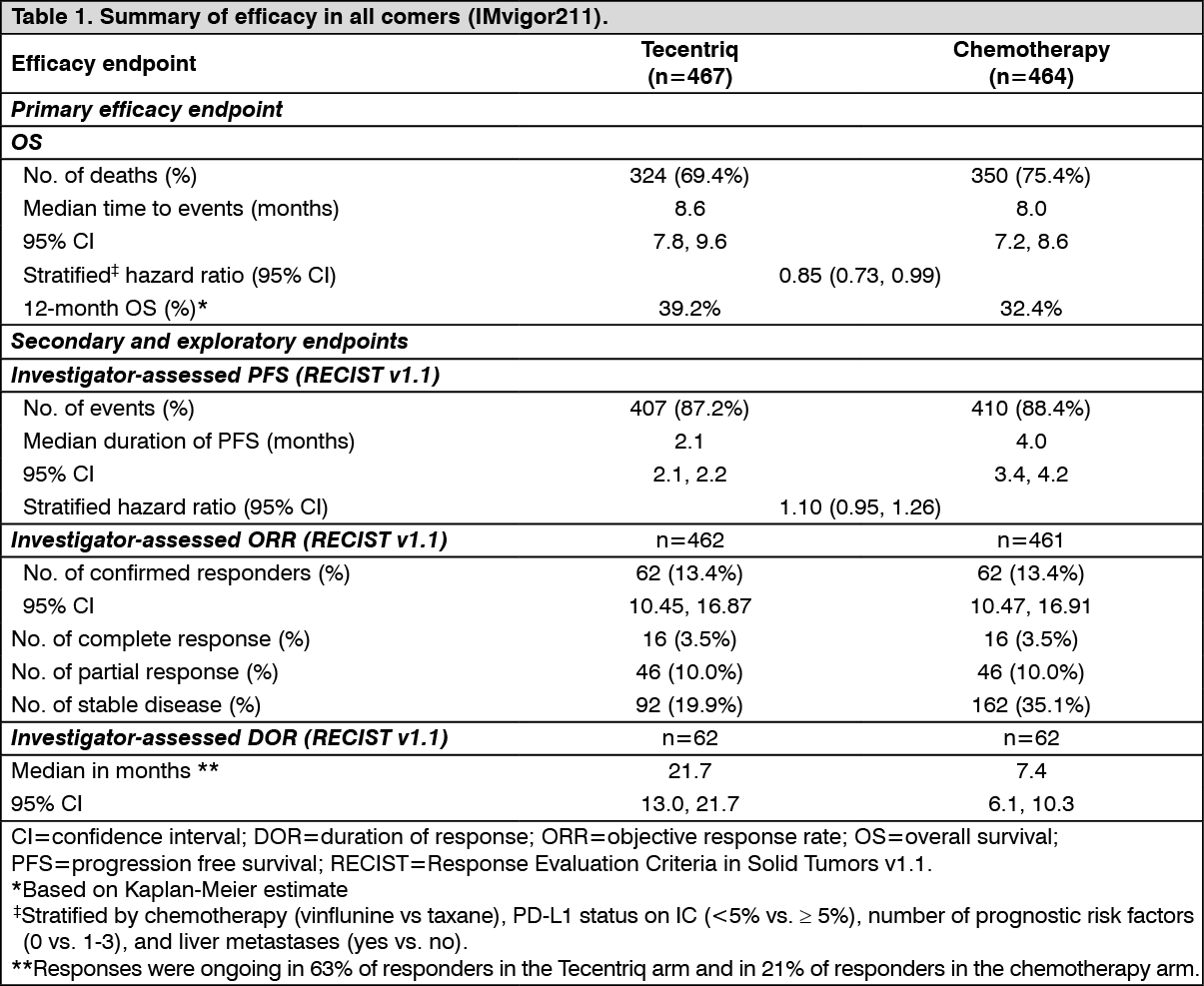
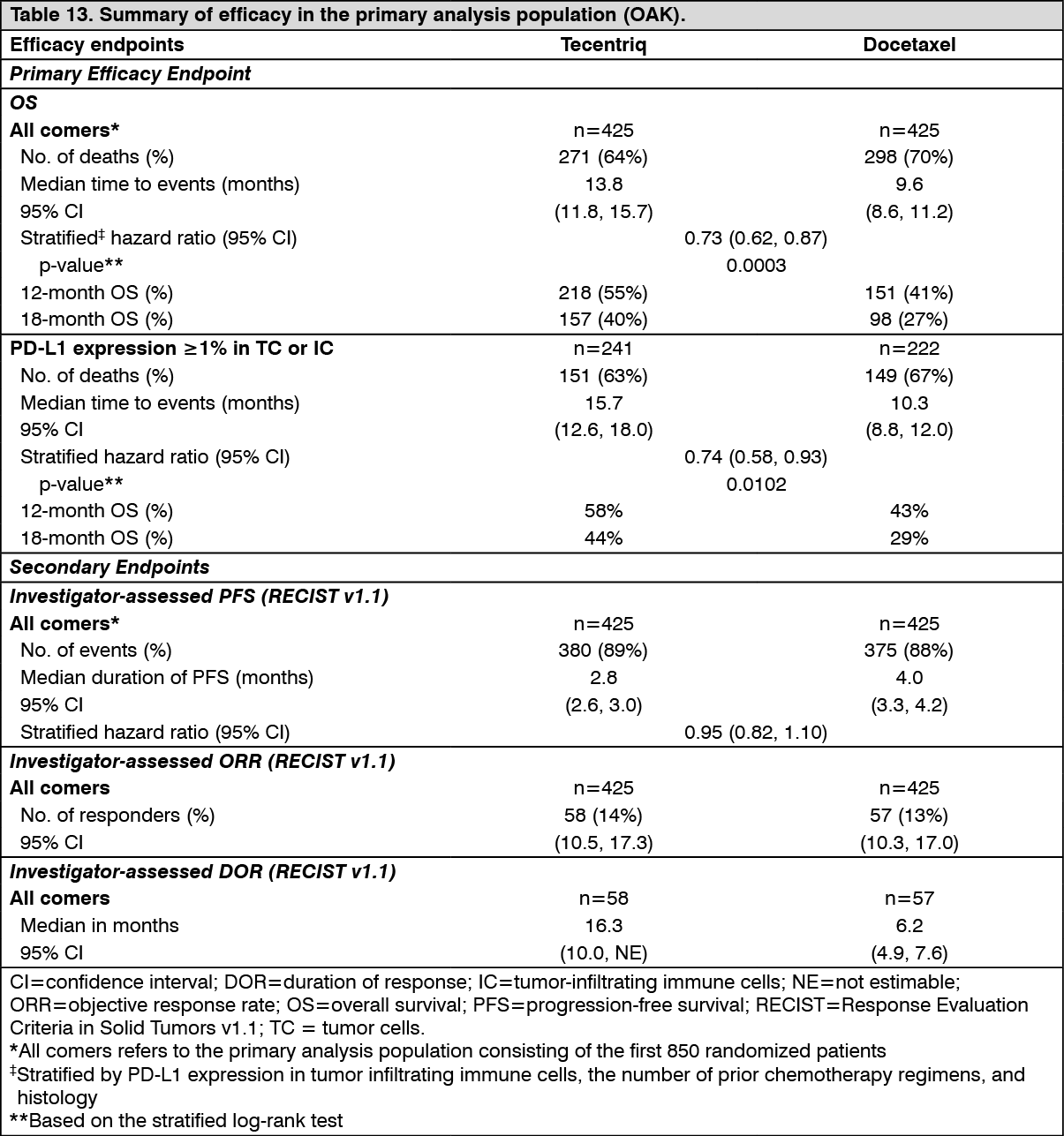
The median survival follow up was 17 months. Study IMvigor211 did not meet its primary endpoint. In the subset of patients with tumors having PD-L1 expression ≥5%, Tecentriq did not demonstrate a statistically significant survival benefit compared to chemotherapy with an OS HR of 0.87 (95% CI: 0.63, 1.21; median OS of 11.1 vs. 10.6 months for Tecentriq and chemotherapy respectively). The stratified log rank p value was 0.41. As a consequence, no formal statistical testing was performed for OS in the PD-L1 expression ≥1% subgroup or in all comers, and results of those analyses are considered exploratory. The key results in the all comer population are summarised in Table 1. The Kaplan Meier curve for OS in the all comer population is presented in Figure 1. (See Table 1 and Figure 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image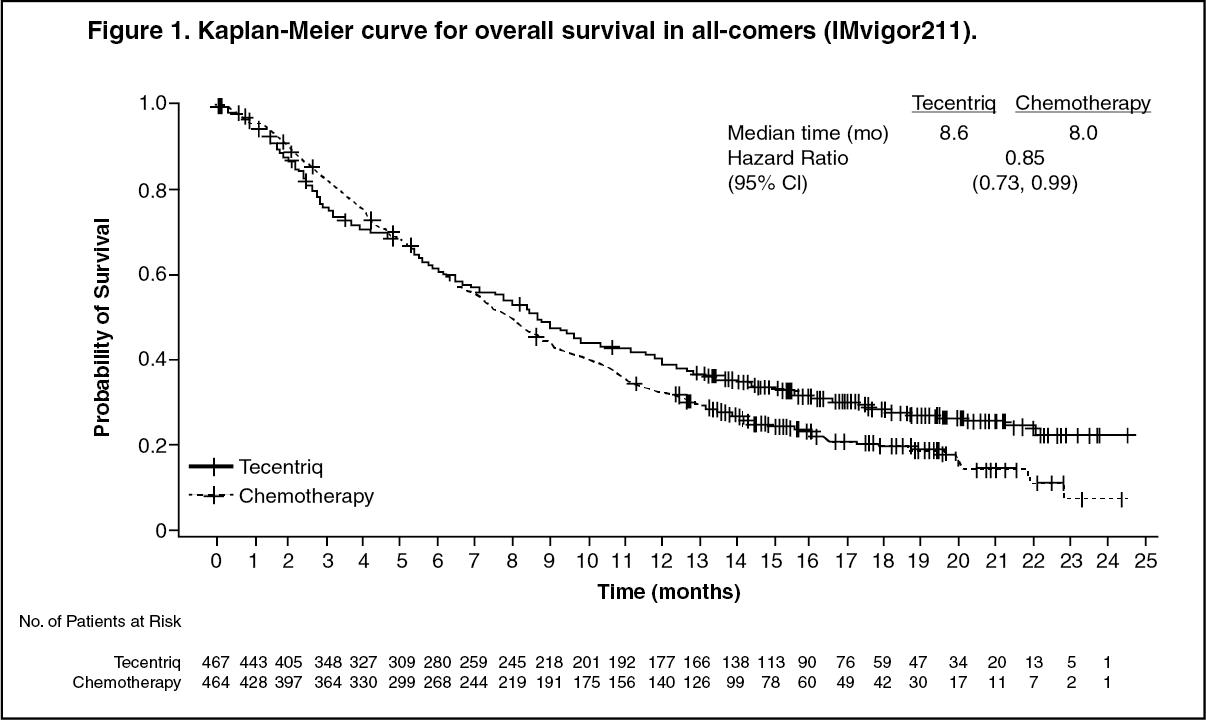 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIMvigor 210: A phase II, multi-center, international, two-cohort, single-arm clinical trial, GO29293 (IMvigor210), was conducted in patients with locally advanced or metastatic urothelial carcinoma (also known as urothelial bladder cancer). The study enrolled a total of 438 patients and had two patient cohorts. Cohort 1 included previously untreated patients with locally advanced or metastatic urothelial carcinoma who were ineligible or unfit for cisplatin-based chemotherapy or had disease progression at least 12 months after treatment with a platinum-containing neoadjuvant or adjuvant chemotherapy regimen. Cohort 2 included patients who received at least one platinum-based chemotherapy regimen for locally advanced or metastatic urothelial carcinoma or had disease progression within 12 months of treatment with a platinum-containing neoadjuvant or adjuvant chemotherapy regimen.
In cohort 1, 119 patients were treated with Tecentriq 1200 mg by intravenous infusion every 3 weeks until disease progression. The median age was 73 years. Most patients were male (81%), and the majority of patients were white (91%).
Cohort 1, included 45 patients (38%) with ECOG performance status of 0, 50 patients (42%) with ECOG performances status of 1 and 24 patients (20%) with ECOG performance status of 2, 35 patients (29%) with no Bajorin risk factors (ECOG performance status ≥2 and visceral metastasis), 66 patients (56%) with one Bajorin risk factor and 18 patients (15%) with two Bajorin risk factors 84 patients (71%) with impaired renal function (GFR < 60 ml/min), and 25 patients (21%) with liver metastasis.
The primary efficacy endpoint for Cohort 1 was confirmed objective response rate (ORR) as assessed by an independent review facility (IRF) using RECIST v1.1. The primary analysis was performed when all patients had at least 24 weeks of follow-up. Median duration of treatment was 15.0 weeks and median duration of survival follow-up was 8.5 months in all comers. Clinically relevant IRF-assessed ORRs per RECIST v1.1 were shown; however, when compared to a prespecified historical control response rate of 10%, statistical significance was not reached for the primary endpoint. The confirmed ORRs per IRF-RECIST v1.1 were 21.9% (95% CI: 9.3, 40.0) in patients with PD-L1 expression ≥ 5%, 18.8% (95% CI: 10.9, 29.0) in patients with PD-L1 expression ≥ 1%, and 19.3% (95% CI: 12.7, 27.6) in all comers. The median duration of response (DOR) was not reached in any PD-L1 expression subgroup or in all comers. OS was not mature with an event ratio of approximately 40%. Median OS for all patient subgroups (PD-L1 expression ≥ 5% and ≥ 1%) and in all comers was 10.6 months.
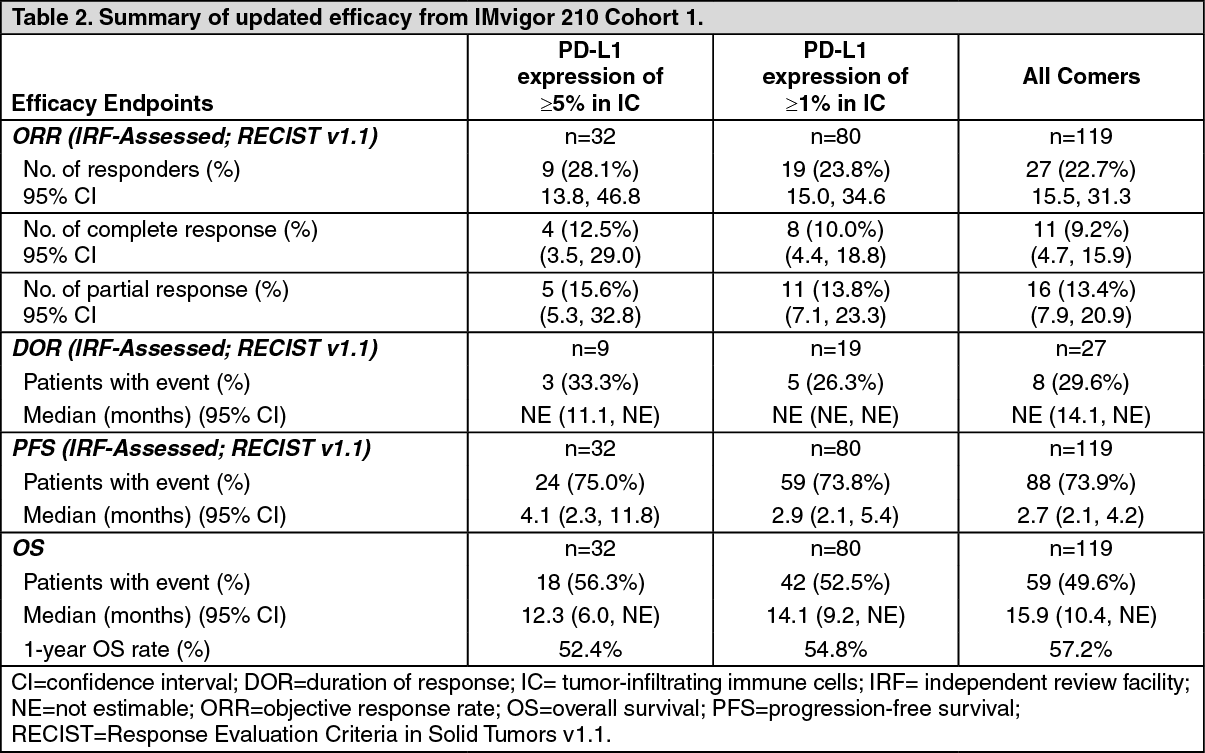
An updated analysis was performed with a median duration of survival follow up of 17.2 months for Cohort 1 and is summarized in Table 2. The median DOR was not reached in any PD-L1 expression subgroup or in all comers. (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn Cohort 2, the co-primary efficacy endpoints were confirmed ORR as assessed by an IRF using RECIST v1.1 and investigator-assessed ORR according to Modified RECIST (mRECIST) criteria. There were 310 patients treated with Tecentriq 1200 mg by intravenous infusion every 3 weeks until loss of clinical benefit. The primary analysis of Cohort 2 was performed when all patients had at least 24 weeks of follow-up. The study met its co-primary endpoints in all subgroups in Cohort 2, demonstrating statistically significant ORRs per IRF-assessed RECIST v1.1 and investigator-assessed mRECIST compared to a prespecified historical control response rate of 10%.
An analysis was also performed with a median duration of survival follow up 21.1 months for Cohort 2. The confirmed ORRs per IRF-RECIST v1.1 were 28.0% (95% CI: 19.5, 37.9) in patients with PD-L1 expression ≥ 5%, 19.3% (95% CI: 14.2, 25.4) in patients with PD-L1 expression ≥ 1%, and 15.8% (95% CI: 11.9, 20.4) in all comers. The confirmed ORR per investigator-assessed mRECIST was 29.0% (95% CI: 20.4, 38.9) in patients with PD-L1 expression ≥ 5%, 23.7% (95% CI: 18.1, 30.1) in patients with PD-L1 expression ≥ 1%, and 19.7% (95% CI: 15.4, 24.6) in all comers. The rate of complete response per IRF-RECIST v1.1 in the all comer population was 6.1% (95% CI: 3.7, 9.4). Median DOR was not reached in any PD-L1 expression subgroups or in all comers, however was reached in patients with PD-L1 expression <1% (13.3 months; 95% CI 4.2, NE). The OS rate at 12 months was 37% in all comers.
NSCLC: Early-stage NSCLC: IMpower010: A phase III, open-label, multi-center, randomized study, GO29527 (IMpower010), was conducted to evaluate the efficacy and safety of Tecentriq for the adjuvant treatment of patients with stage IB (tumors ≥ 4 cm) - IIIA NSCLC (per the Union for International Cancer Control/American Joint Committee on Cancer staging system, 7th edition). A total of 1280 enrolled patients had complete tumor resection and were eligible to receive up to 4 cycles of cisplatin-based chemotherapy. The cisplatin-based chemotherapy regimens are described in Table 3. (See Table 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAfter completion of cisplatin-based chemotherapy (up to four cycles), a total of 1005 patients were randomized in a 1:1 ratio to receive Tecentriq (Arm A) or best supportive care (BSC) (Arm B). Tecentriq was administered as a fixed dose of 1200 mg by IV infusion every 3 weeks for 16 cycles unless there was disease recurrence or unacceptable toxicity. Randomization was stratified by sex, stage of disease, histology, and PD-L1 expression.
Patients were excluded if they had a history of autoimmune disease; administration of a live, attenuated vaccine within 28 days prior to randomization; administration of systemic immunostimulatory agents within 4 weeks or systemic immunosuppressive medications within 2 weeks prior to randomization. Tumor assessments were conducted at baseline of the randomization phase and every 4 months for the first year following Cycle 1, Day 1 and then every 6 months until year five, then annually thereafter.
The demographics and baseline disease characteristics were well balanced between the treatment arms. The median age was 62 years (range: 26 to 84), and 67% of patients were male. The majority of patients were White (73%), and 24% were Asian. Most patients were current or previous smokers (78%) and baseline ECOG performance status in patients was 0 (55%) or 1 (44%). Overall, 12% of patients had stage IB, 47% had stage II and 41% had stage IIIA disease. The percentage of patients who had tumors with PD-L1 expression ≥ 1% on TC as measured by the VENTANA PD-L1 (SP263) Assay was 55%.
The primary efficacy outcome measure was disease-free survival (DFS) as assessed by the investigator. DFS was defined as the time from the date of randomization to the date of occurrence of any of the following: first documented recurrence of disease, new primary NSCLC, or death due to any cause, whichever occured first. A key secondary efficacy outcome measure was overall survival (OS).
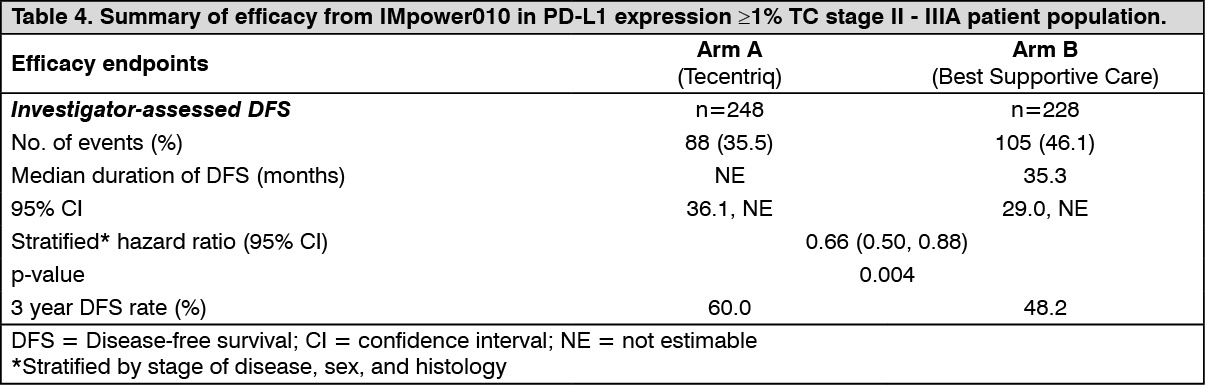
At the time of the interim DFS analysis, the study met its primary endpoint and demonstrated a statistically significant and clinically meaningful improvement in DFS in the Tecentriq arm compared to the BSC arm in the PD-L1 ≥ 1% TC stage II - IIIA patient population. The median follow-up time was approximately 32 months. The OS data were immature at the time of the DFS interim analysis with approximately 18.9% of deaths reported in both arms in the PD-L1 ≥ 1% TC stage II - IIIA patient population. An exploratory analysis of OS suggested a trend in favor of Tecentriq over BSC (stratified HR=0.77 [95% CI: 0.51, 1.17]) in this patient population.
The study also demonstrated a statistically significant improvement in DFS for all randomized stage II-IIIA patients (stratified HR: 0.79, [95% CI 0.64, 0.96], p-value 0.0205).
The key efficacy results are summarized in Table 4. The Kaplan-Meier curve for DFS is presented in Figure 2. (See Table 4 and Figure 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image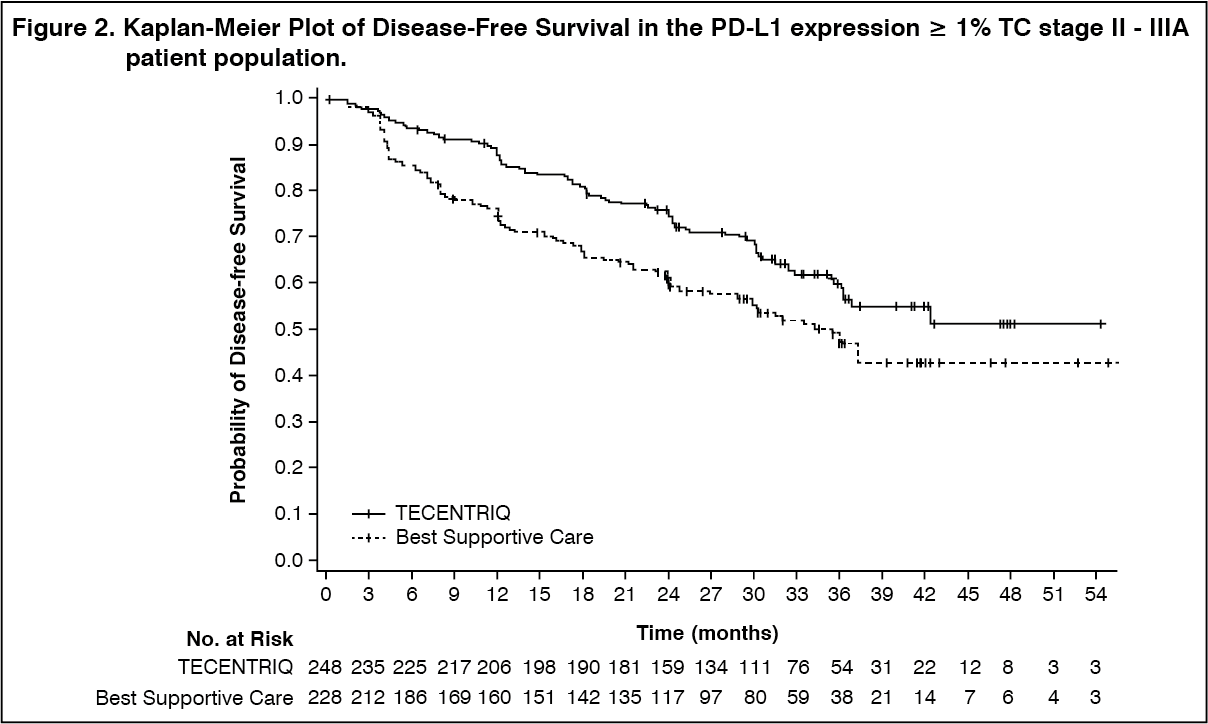 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe observed DFS improvement in the Tecentriq arm compared with the BSC arm was consistently shown across the majority of pre-specified subgroups in the PD-L1 ≥ 1% TC stage II - IIIA patient population including both non-squamous NSCLC patients (unstratified HR: 0.60 [95% CI: 0.42, 0.84], median DFS 42.3 vs. 30.1 months) and squamous NSCLC patients (unstratified HR: 0.78 [95% CI: 0.47, 1.29], median DFS (NE vs. NE months).
1L metastatic non-squamous NSCLC: IMpower 150: A phase III, open-label, randomized study, GO29436 (IMpower150), was conducted to evaluate the efficacy and safety of Tecentriq in combination with paclitaxel and carboplatin, with or without bevacizumab, in chemotherapy-naïve patients with metastatic non-squamous NSCLC. A total of 1202 patients were enrolled and were randomized in a 1:1:1 ratio to receive one of the treatment regimens described in Table 5. Randomization was stratified by sex, presence of liver metastases and PD-L1 tumor expression on tumor cells (TC) and tumor infiltrating cells (IC). (See Table 5.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePatients were excluded if they had history of autoimmune disease; administration of a live, attenuated vaccine within 28 days prior to randomization; administration of systemic immunostimulatory agents within 4 weeks or systemic immunosuppressive medications within 2 weeks prior to randomization; active or untreated CNS metastases; clear tumor infiltration into the thoracic great vessels or clear cavitation of pulmonary lesions, as seen on imaging. Tumor assessments were conducted every 6 weeks for the first 48 weeks following Cycle 1, Day 1 and then every 9 weeks thereafter.
The demographics and baseline disease characteristics of the study population were well balanced between the treatment arms. In this study, the median age was 63 years (range: 31 to 90), and 60% of patients were male. The majority of patients were white (82%). Approximately 10% of patients had known EGFR mutations, 4% had known ALK rearrangements, 14% had liver metastases at baseline, and most patients were current or previous smokers (80%). Baseline ECOG performance status was 0 (43%) or 1 (57%).
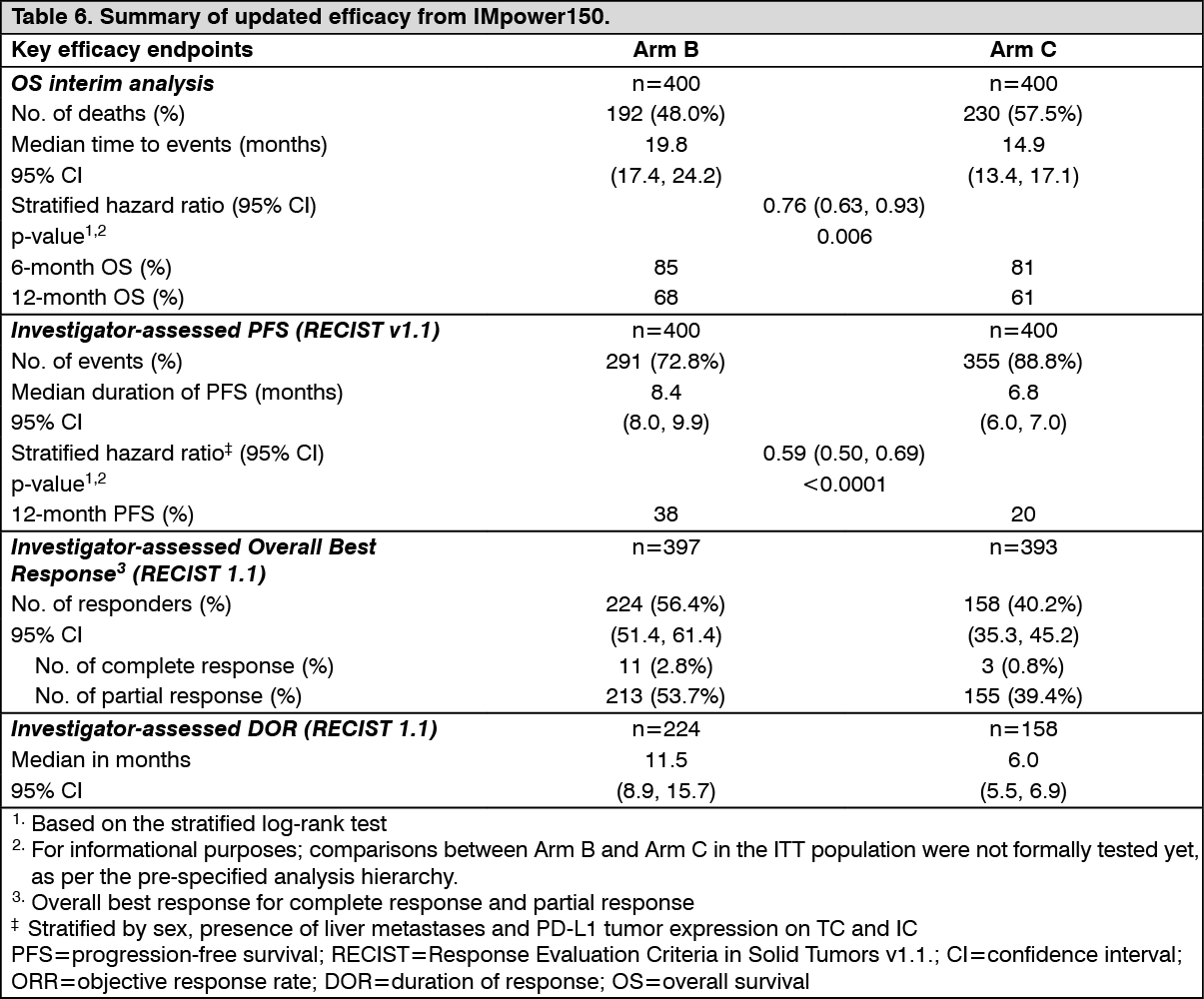
At the time of the final analysis for PFS, patients had a median follow up time of 15.3 months. The ITT population included patients with EGFR mutations or ALK rearrangements who should have been previously treated with tyrosine kinase inhibitors, demonstrated PFS improvement in Arm B as compared to Arm C (HR: 0.61 [95% CI: 0.52, 0.72] median PFS 8.3 vs 6.8 months).
At the time of the interim OS analysis, patients had a median follow up time of 19.7 months. The key results from this analysis are summarized in Table 6. Kaplan-Meier curves for OS in the ITT population are presented in Figure 3. Figure 4 summarizes the results of OS in the ITT and PD-L1 subgroups, demonstrating OS benefit with Tecentriq in all subgroups, including those with PD-L1 expression <1% on TC and IC. Updated PFS results are also demonstrated in Figure 5 and 6. (See Table 6, Figures 3, 4, 5 and 6.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image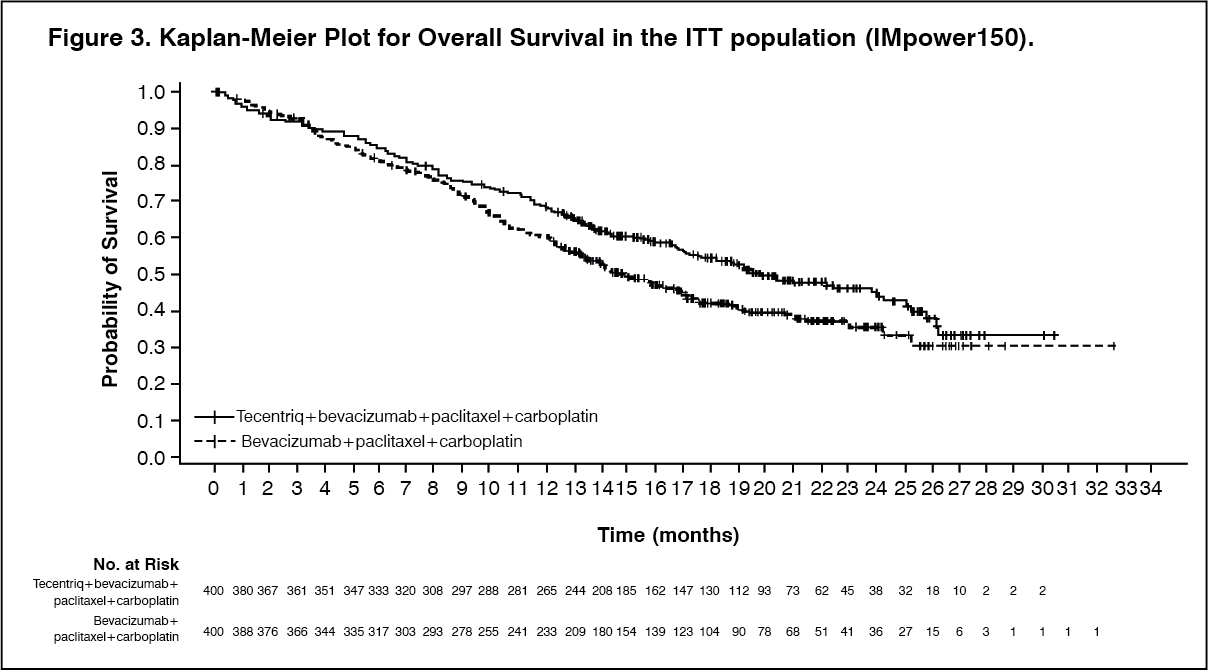 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image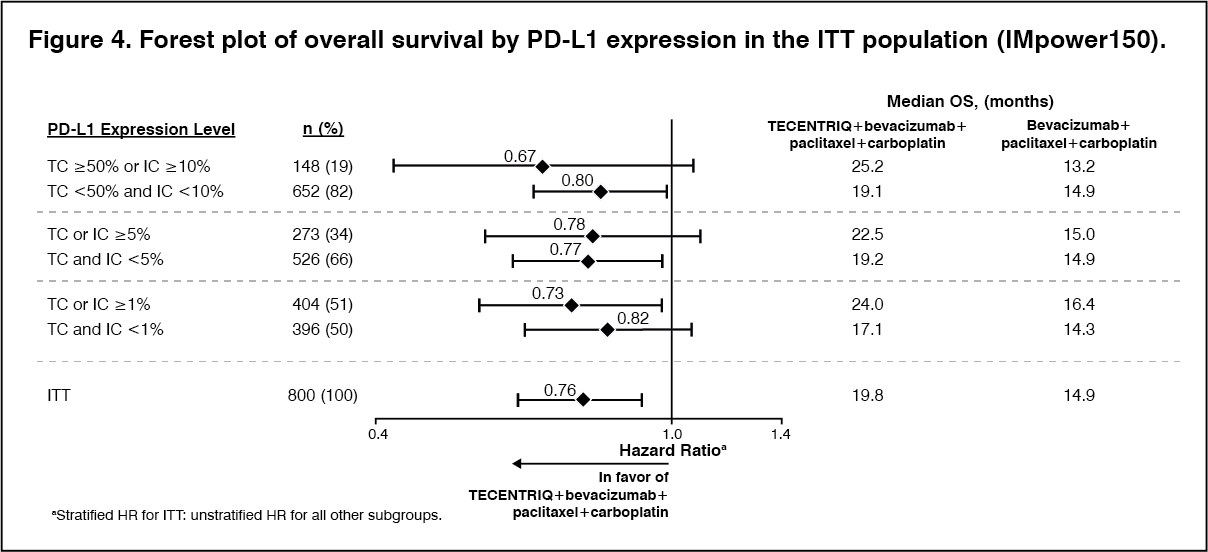 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image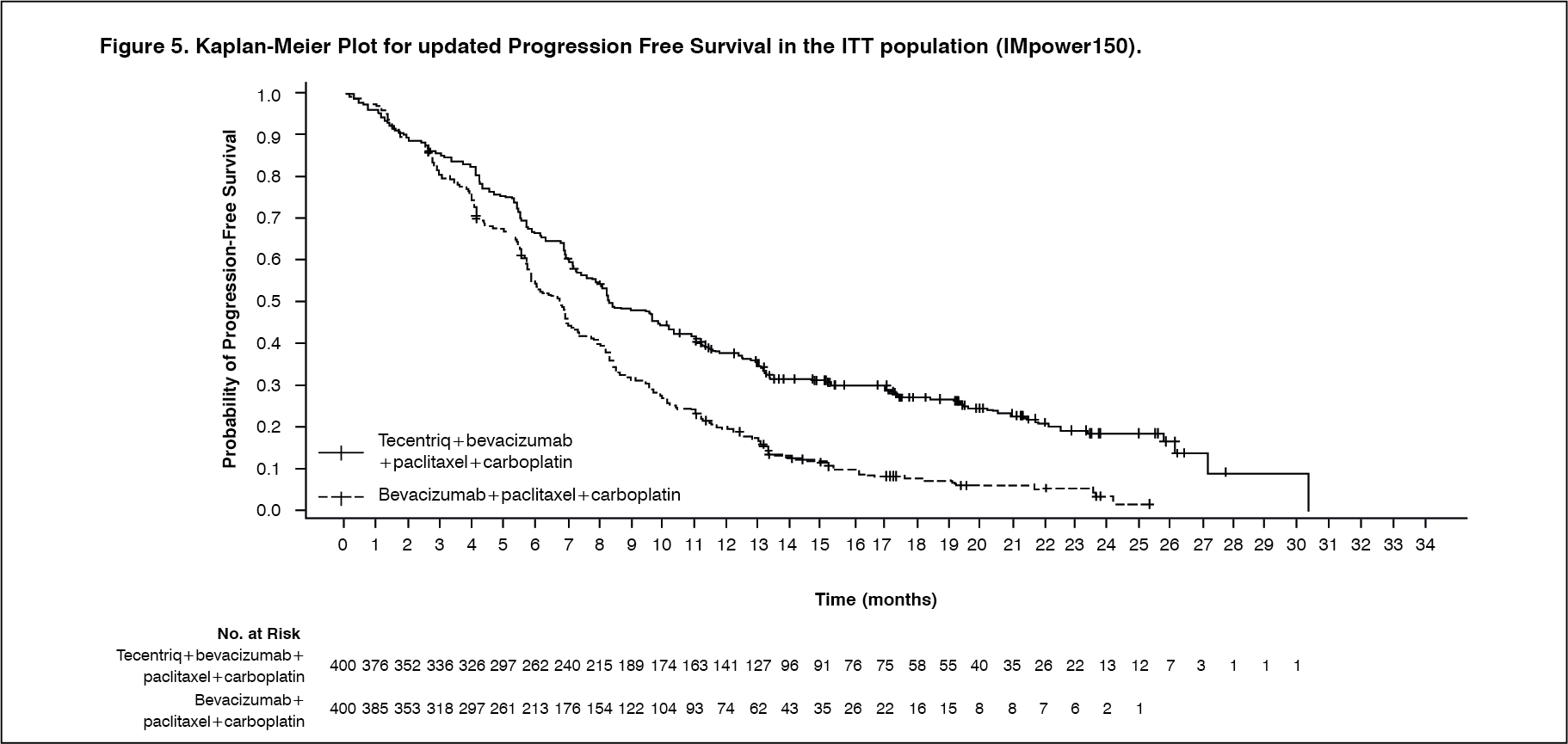 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image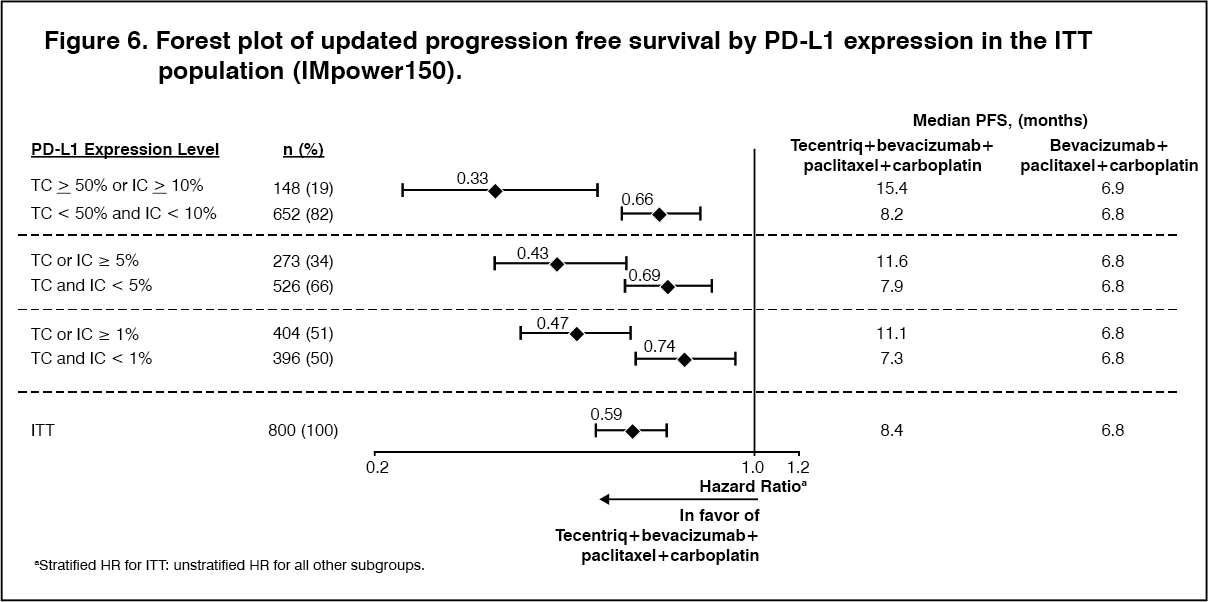 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePre-specified subgroup analyses from the interim OS analysis showed numerical OS improvements in the Tecentriq with bevacizumab, paclitaxel, carboplatin arm as compared to the bevacizumab, paclitaxel and carboplatin arm for patients with EGFR mutations or ALK rearrangements (HR: 0.54 [95% CI: 0.29, 1.03], median OS NE vs. 17.5 months) and liver metastases (HR: 0.52 [95% CI: 0.33, 0.82], median OS 13.3 vs 9.4 months). Numerical PFS improvements were also shown in patients with EGFR mutations or ALK rearrangements (HR: 0.55 [95% CI 0.34, 0.90], median PFS 10 vs. 6.1 months) and liver metastases (HR: 0.41 [95% CI 0.26, 0.62], median PFS 8.2 vs. 5.4 months).
This study also evaluated Physical Function and Patient-Reported Treatment-Related Symptoms using the EORTC QLQ-C30 and EORTC QLQ-LC13 measures at the time of the final PFS analysis. On average, patients who received Tecentriq with bevacizumab, paclitaxel and carboplatin reported minimal treatment burden as indicated by minimal deterioration in both Physical Function and Patient-Reported Treatment-Related Symptom Scores (i.e. fatigue, constipation, diarrhea, nausea/vomiting, hemoptysis, dysphagia, and sore mouth) while on treatment. Average patient-reported physical function and treatment-related symptom scores in both patients who received Tecentriq with bevacizumab, paclitaxel and carboplatin as well as patients who received bevacizumab in combination with paclitaxel and carboplatin, were comparable while on treatment.
IMpower130: A Phase III, open-label, randomized study, GO29537 (IMpower130) was conducted to evaluate the efficacy and safety of Tecentriq in combination with nab-paclitaxel and carboplatin, in chemotherapy-naïve patients with metastatic non-squamous NSCLC. Patients including those with EGFR or ALK genomic tumor aberrations, were enrolled and were randomized in a 2:1 ratio to receive one of the treatment regimens described in Table 7. Randomization was stratified by sex, presence of liver metastases and PD-L1 tumor expression on tumor cells (TC) and tumor infiltrating cells (IC). Patients in treatment regimen B were able to crossover and receive Tecentriq monotherapy following disease progression. (See Table 7.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePatients were excluded if they had history of autoimmune disease, administration of live, attenuated vaccine within 28 days prior to randomization, administration of immunostimulatory agents within 4 weeks or systemic immunosuppressive medications within 2 weeks prior to randomization, and active or untreated CNS metastases. Tumor assessments were conducted every 6 weeks for the first 48 weeks following Cycle 1, then every 9 weeks thereafter.
The demographics and baseline disease characteristics of the study population (n = 723) were well balanced between the treatment arms. The median age was 64 years (range 18 to 86). The majority of the patients were, male (57%), white (90%). 14.8% of patients had liver metastases at baseline, and most patients were current or previous smokers (88%). The majority of patients had baseline ECOG performance status of 1 (58.7%).
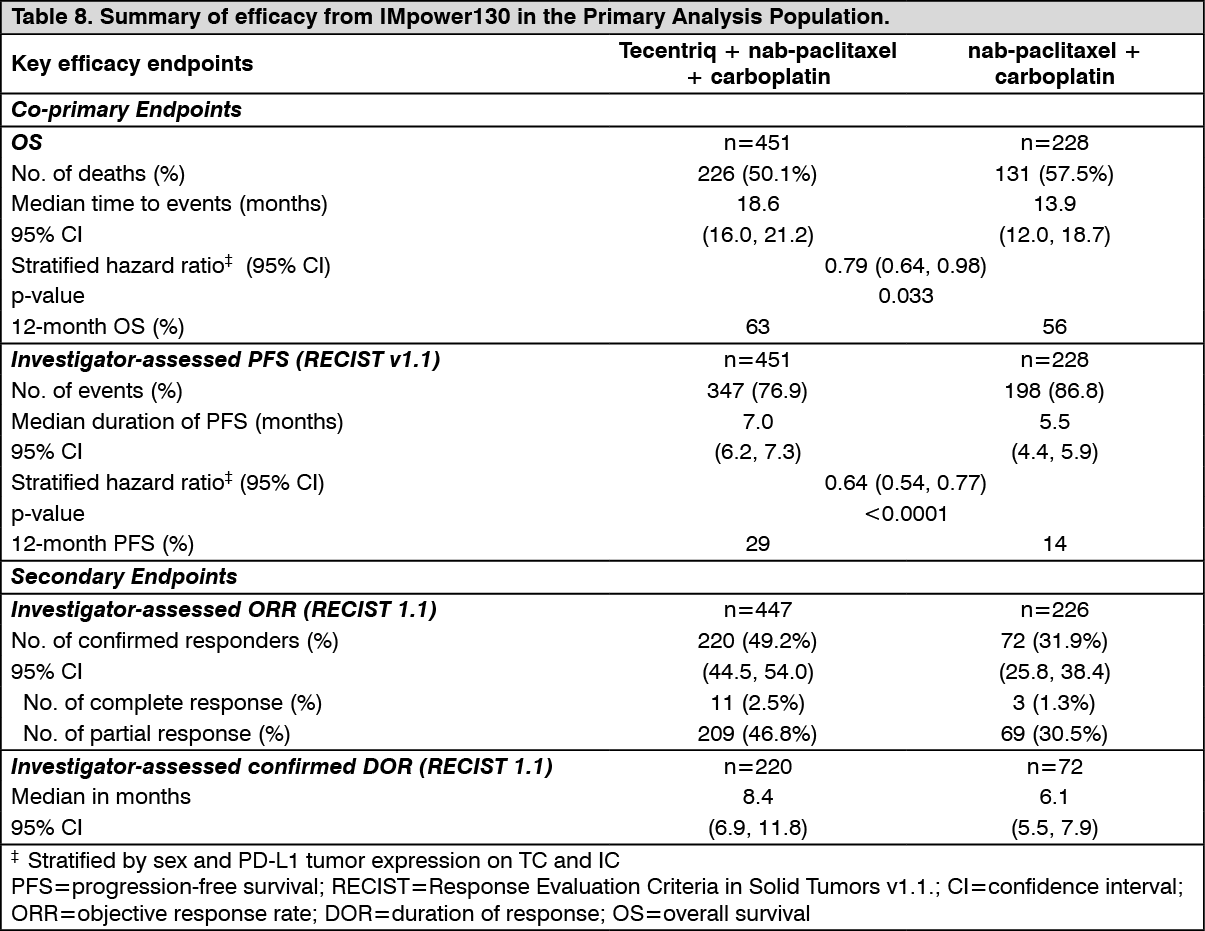
The primary analysis was conducted in all patients, excluding those with EGFR or ALK genomic tumor aberrations (n = 679). Patients had a median survival follow up time of 18.6 months. Improvements in OS and PFS were demonstrated with Tecentriq + nab-paclitaxel + carboplatin compared to the control. The key results are summarized in Table 8 and Kaplan-Meier curves for OS and PFS are presented in Figures 7 and 9, respectively.
All PD-L1 subgroups, regardless of expression, derived benefit in terms of OS and PFS; the results are summarized in Figure 8 and 10. Consistent OS and PFS benefit was demonstrated in all other pre-specified subgroups, with the exception of patients with liver metastases who did not show improved OS with Tecentriq, nab-paclitaxel and carboplatin, compared to nab-paclitaxel and carboplatin (HR of 1.04, 95% CI: 0.63,1.72).
Approximately 66% of patients in the nab-paclitaxel and carboplatin arm received any anti-cancer therapy after disease progression compared to 39% in the Tecentriq, nab-paclitaxel and carboplatin arm. These included, approximately 59% of patients in the nab-paclitaxel and carboplatin arm received any cancer immunotherapy after disease progression, which includes Tecentriq as crossover (41% of all patients), compared to 7.3% in the Tecentriq, nab-paclitaxel and carboplatin arm. (See Table 8, Figures 7, 8, 9 and 10.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image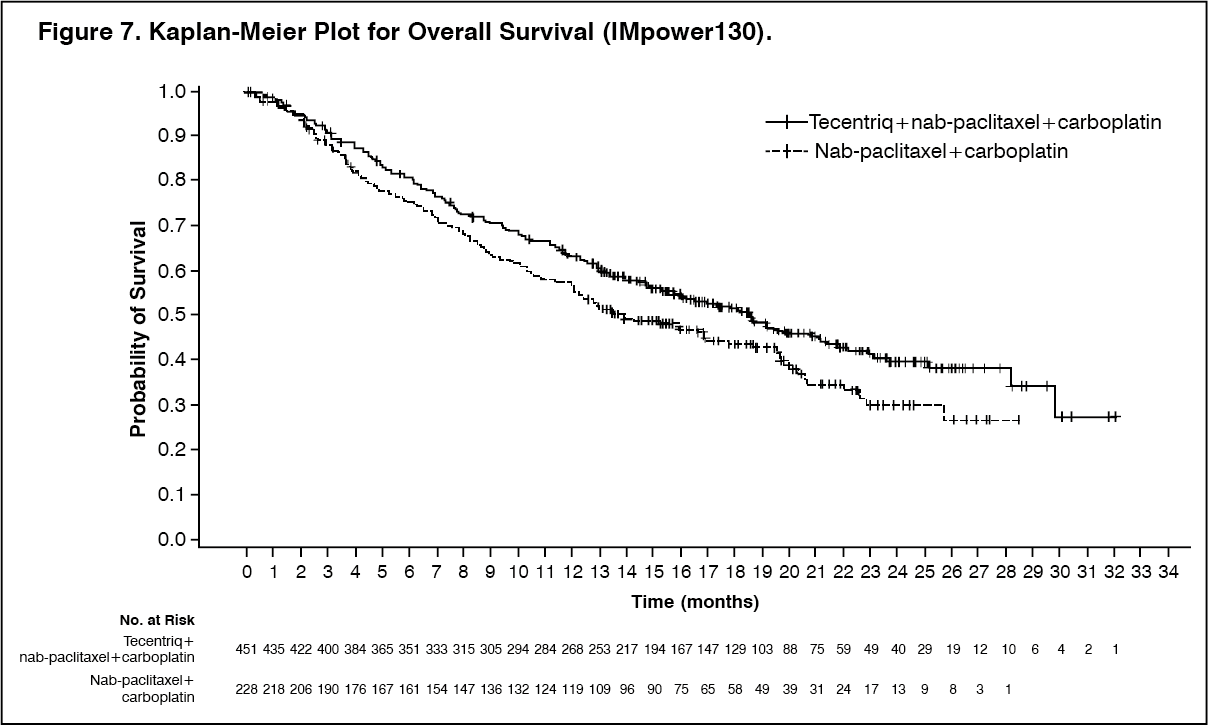 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image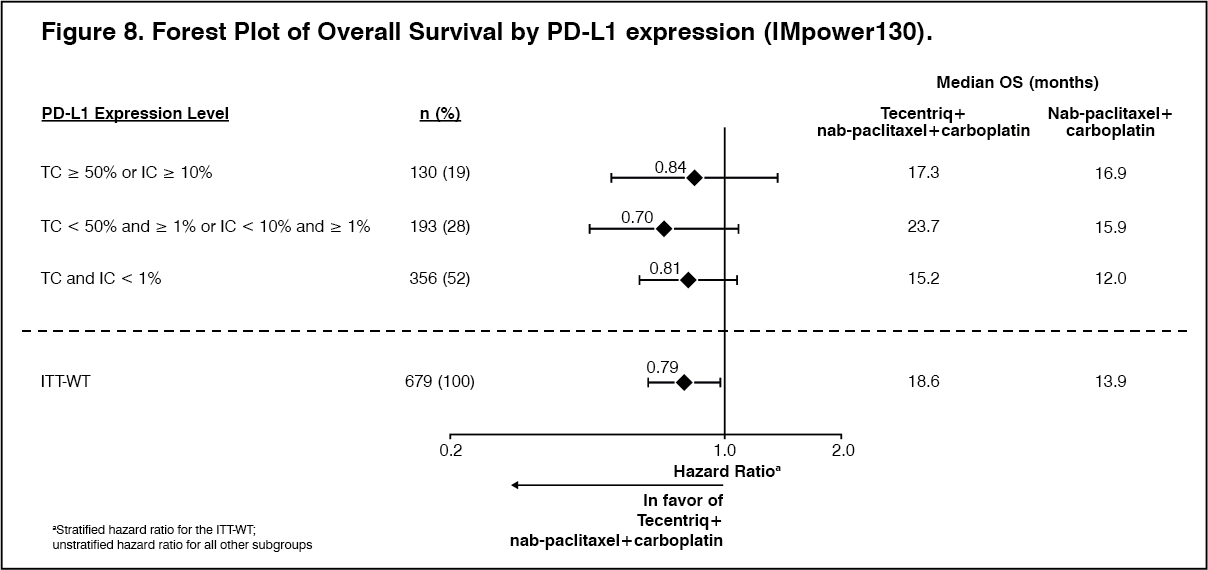 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image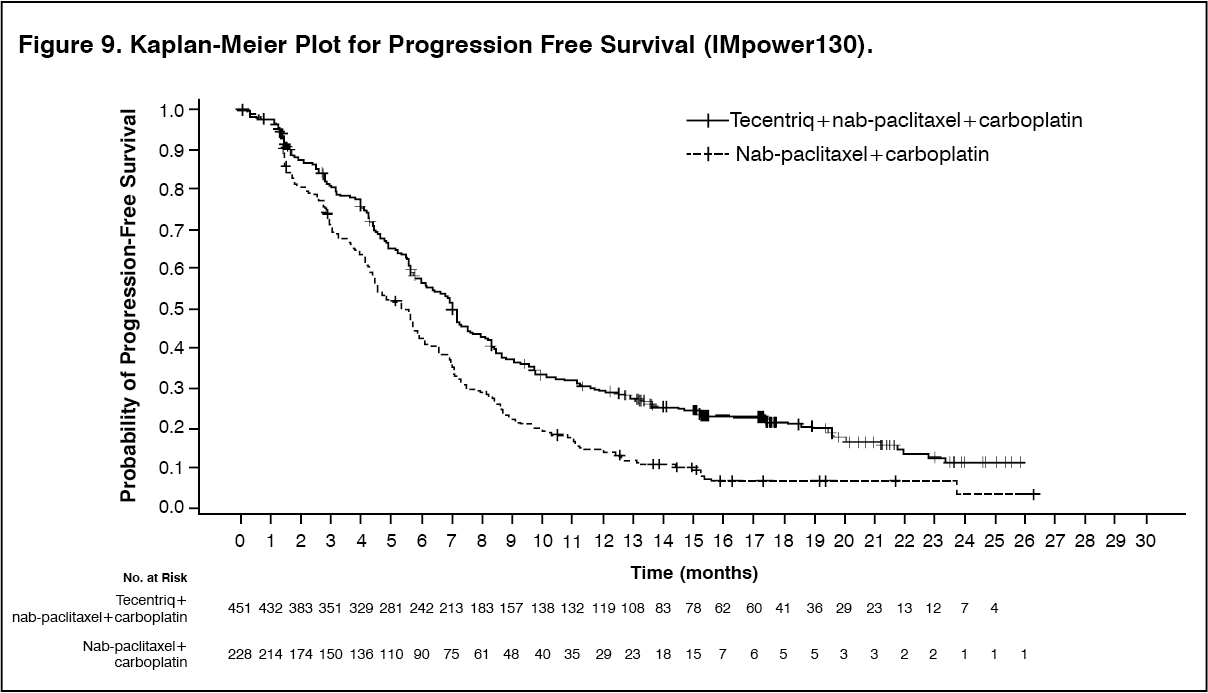 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image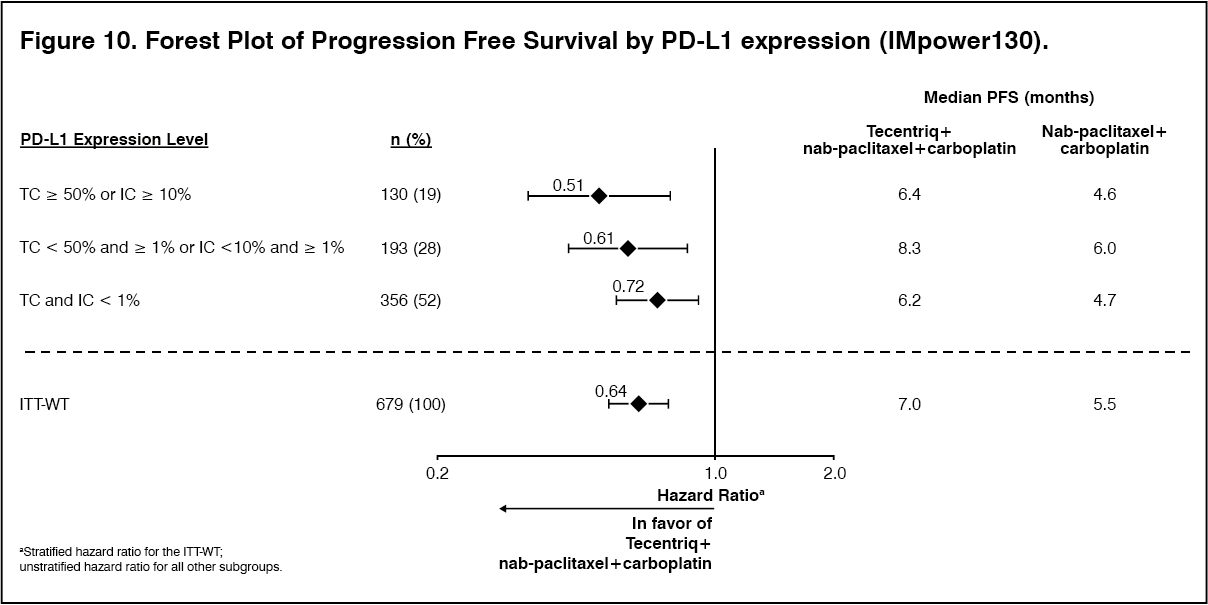 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe study also evaluated Physical Function and Patient Reported Treatment-Related Symptoms using the EORTC QLQ-C30 and EORTC QLQ-LC13 measures. On average, patients who received Tecentriq with nab-paclitaxel and carboplatin reported high functioning and no clinically meaningful worsening in treatment-related symptoms. There was no difference in delay of lung-related symptoms (dyspnea, cough and chest pain) however patients receiving Tecentriq, nab-paclitaxel and carboplatin reported less worsening of these symptoms over time.
1L metastatic non-squamous and squamous NSCLC: IMpower110: A phase III, open-label, multi-center, randomized study, GO29431 (IMpower110), was conducted to evaluate the efficacy and safety of Tecentriq in chemotherapy-naïve patients with metastatic NSCLC, with PD-L1 expression ≥ 1% TC (PD-L1 stained ≥ 1% of tumor cells) or ≥ 1% IC (PD-L1 stained tumor-infiltrating immune cells covering ≥ 1% of the tumor area) by the VENTANA PD-L1 (SP142) Assay.
A total of 572 patients were randomized in a 1:1 ratio to receive Tecentriq (Arm A) or chemotherapy (Arm B). Tecentriq was administered as a fixed dose of 1200 mg by IV infusion every 3 weeks until loss of clinical benefit as assessed by the investigator or unacceptable toxicity. The chemotherapy regimens are described in Table 9. Randomization was stratified by sex, ECOG performance status, histology, and PD-L1 tumor expression on TC and IC. (See Table 9.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePatients were excluded if they had history of autoimmune disease; administration of a live, attenuated vaccine within 28 days prior to randomization; administration of systemic immunostimulatory agents within 4 weeks or systemic immunosuppressive medications within 2 weeks prior to randomization; active or untreated CNS metastases. Tumor assessments were conducted every 6 weeks for the first 48 weeks following Cycle 1, Day 1 and then every 9 weeks thereafter.
The demographics and baseline disease characteristics in patients with PD-L1 expression ≥ 1% TC or ≥ 1% IC who do not have EGFR or ALK genomic tumor aberrations (n=554) were well balanced between the treatment arms. The median age was 64.5 years (range: 30 to 87), and 70% of patients were male. The majority of patients were white (84%) and Asian (14%). Most patients were current or previous smokers (87%) and baseline ECOG performance status in patients was 0 (36%) or 1 (64%). Overall, 69% of patients had non-squamous disease and 31% of patients had squamous disease. The demographics and baseline disease characteristics in patients with high PD-L1 expression (PD-L1 ≥ 50% TC or ≥ 10% IC) who do not have EGFR or ALK genomic tumor aberrations (n=205) were generally representative of the broader study population and were balanced between the treatment arms.
The primary endpoint was overall survival (OS). At the time of the interim OS analysis, patients with high PD-L1 expression excluding those with EGFR or ALK genomic tumor aberrations (n=205) demonstrated statistically significant improvement in OS for the patients randomized to Tecentriq (Arm A) as compared with chemotherapy (Arm B). The median survival follow-up time in patients with high PD-L1 expression was 15.7 months. The key results are summarized in Table 10. The Kaplan-Meier curves for OS and PFS in patients with high PD-L1 expression are presented in Figure 11 and 12. (See Table 10, Figures 11 and 12.)
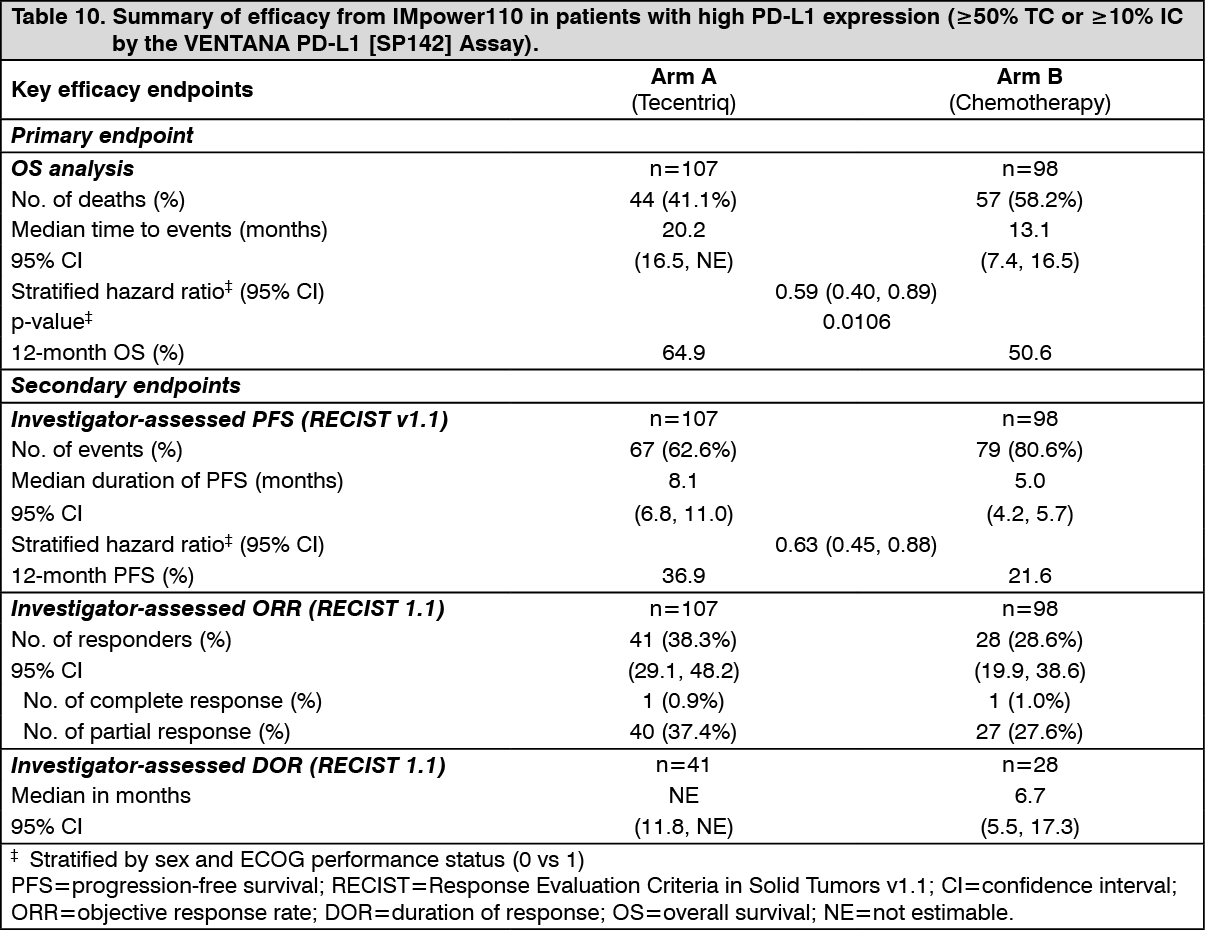 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image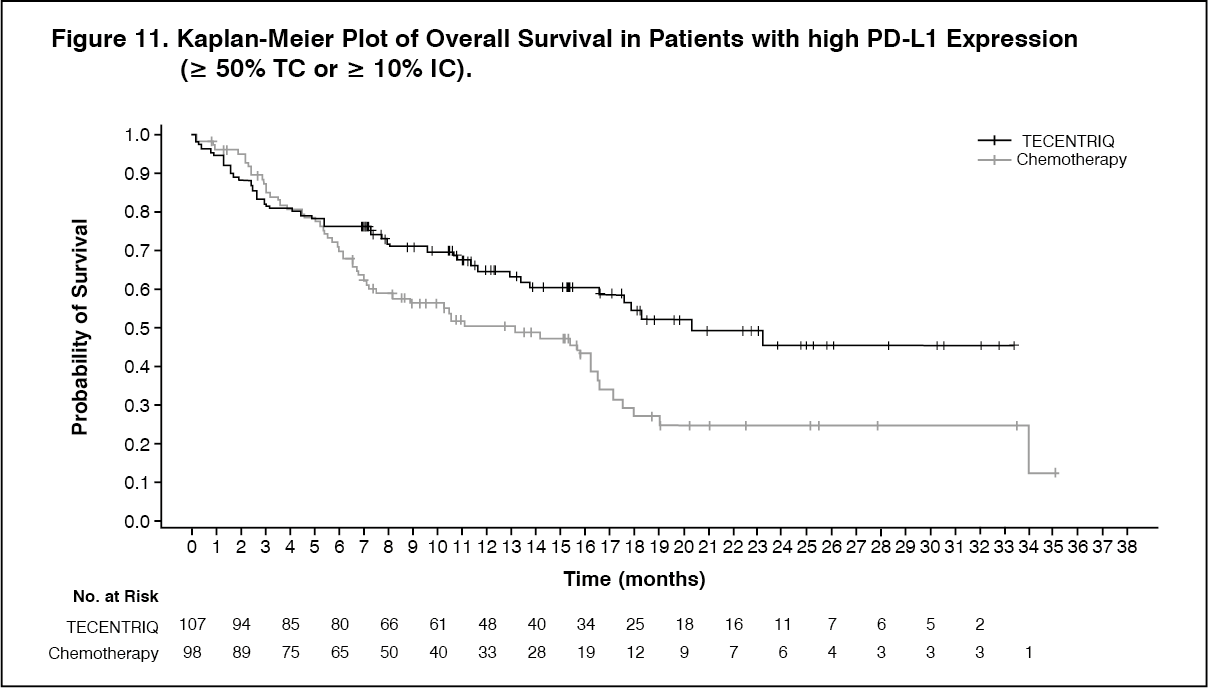 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe observed OS improvement in the Tecentriq arm compared with the chemotherapy arm was consistently demonstrated across subgroups in patients with high PD-L1 expression including both non-squamous NSCLC patients (HR: 0.62 [95% CI: 0.40, 0.96], median OS 20.2 vs. 10.5 months) and squamous NSCLC patients (HR: 0.56 [95% CI: 0.23, 1.37]) median OS NE vs 15.3 months). The data for patients ≥75 years old and patients who were never smokers are too limited to draw conclusions in these subgroups.
Additional pre-specified analyses were conducted to evaluate efficacy by PD-L1 status assessed by the VENTANA PD-L1 (SP263) Assay and by the PD-L1 IHC 22C3 pharmDxTM kit in all randomized patients with PD-L1 expression ≥ 1% TC or ≥ 1% IC by the VENTANA PD-L1 (SP142) Assay who do not have EGFR or ALK genomic tumour abberations (n=554). An OS improvement was observed with atezolizumab compared to chemotherapy in patients with high PD-L1 expression (PD-L1 ≥50% TC) using the VENTANA PD-L1 (SP263) Assay (n=293; HR: 0.71 [95% CI: 0.50, 1.00], median OS 19.5 vs. 16.1 months) and in patients with high PD-L1 expression (Tumour Proportion Score (TPS) ≥50%) using the PD-L1 IHC 22C3 pharmDxTM Kit (n=260; HR: 0.60 [95% CI: 0.42, 0.86], median OS 20.2 vs 11.0 months).
The study also evaluated Patient Reported Physical Function, Global Health Status/Health Related Quality of Life and Lung Related Symptoms using the EORTC QLQ-C30, EORTC QLQ-LC13, and SILC measures at the time of interim OS analysis. Patients who were randomized to Tecentriq (Arm A) on average reported sustained moderate improvement in physical functioning and no worsening in lung cancer-related symptoms (dyspnea, cough, and chest pain) compared to patients randomized to chemotherapy (Arm B). Time to deterioration of these lung-related symptoms as measured by the SILC and EORTC QLQ-LC13 was similar in both treatment groups indicating that patients maintained low disease burden for a comparable duration of time.
1L ES-SCLC: IMpower133: A Phase I/III, randomized, multicenter, double-blind, placebo controlled study, GO30081 (IMpower133), was conducted to evaluate the efficacy and safety of Tecentriq in combination with carboplatin and etoposide in patients with chemotherapy-naïve ES-SCLC. A total of 403 patients were randomized (1:1) to receive one of the treatment regimens described in Table 11. Randomization was stratified by sex, ECOG performance status, and presence of brain metastases.
This study excluded patients who had active or untreated CNS metastases; history of autoimmune disease; administration of live, attenuated vaccine within 4 weeks prior to randomization; administration of systemic immunosuppressive medications within 1 week prior to randomization. Tumor assessments were conducted every 6 weeks for the first 48 weeks following Cycle 1, Day 1 and then every 9 weeks thereafter. Patients treated beyond disease progression had tumor assessment conducted every 6 weeks until treatment discontinuation. (See Table 11.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe demographic and baseline disease characteristics of the primary analysis population were well balanced between the treatment arms. The median age was 64 years (range: 26 to 90 years). The majority of patients were male (65%), white (80%), and 9% had brain metastases and most patients were current or previous smokers (97%). Baseline ECOG performance status was 0 (35%) or 1 (65%).
At the time of the primary analysis, patients had a median survival follow up time of 13.9 months. The key results are summarized in Table 12. Kaplan-Meier curves for OS and PFS are presented in Figure 13 and 14. (See Table 12, Figures 13 and 14.)
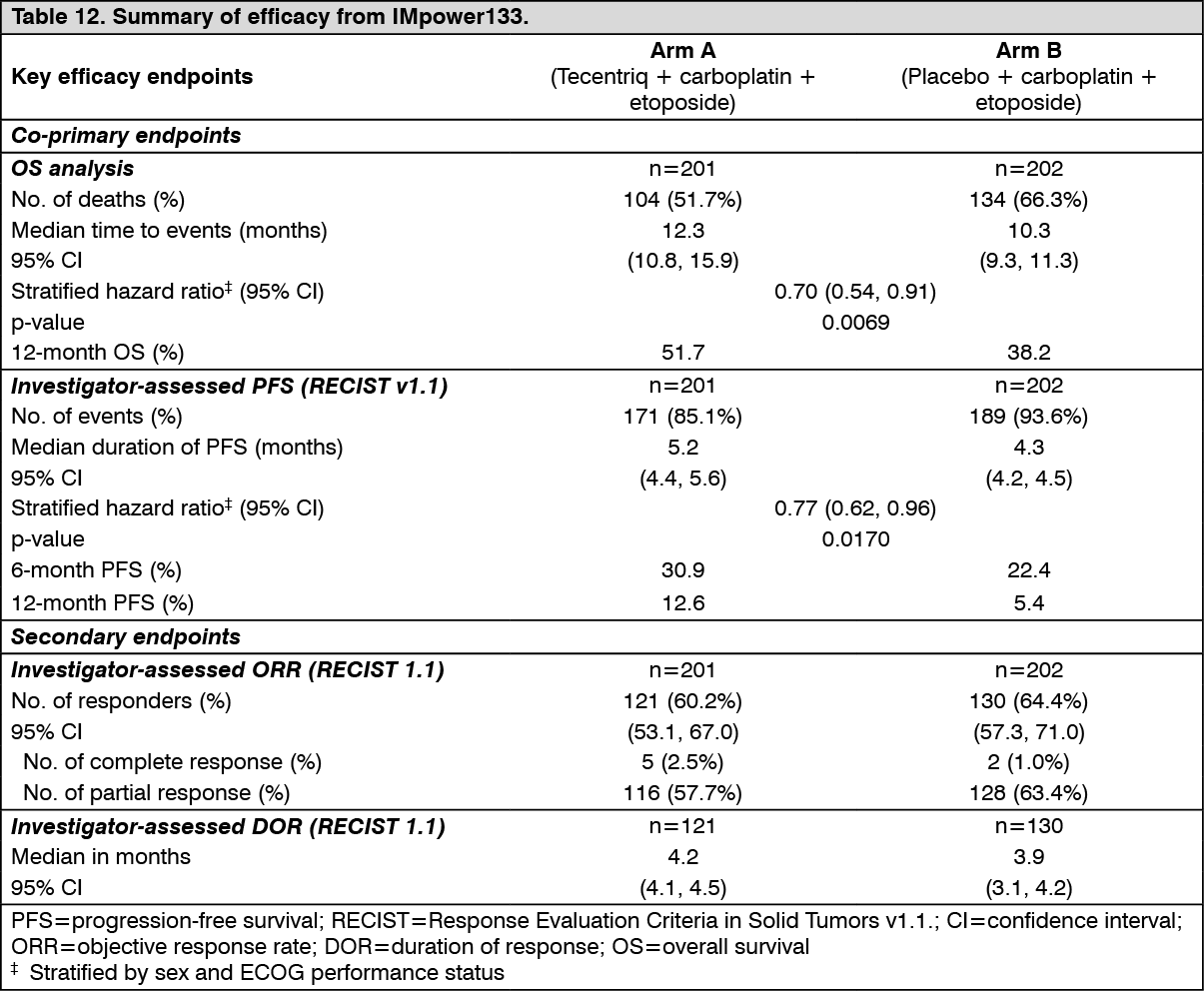 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image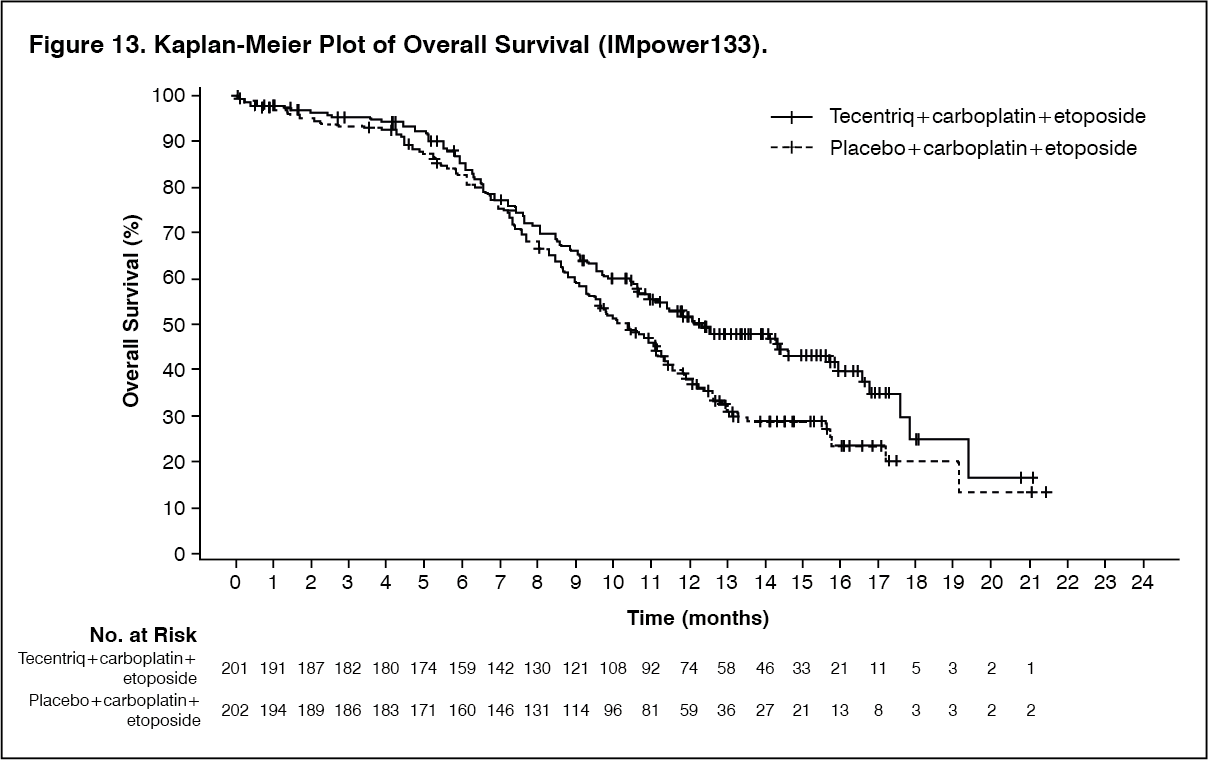 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image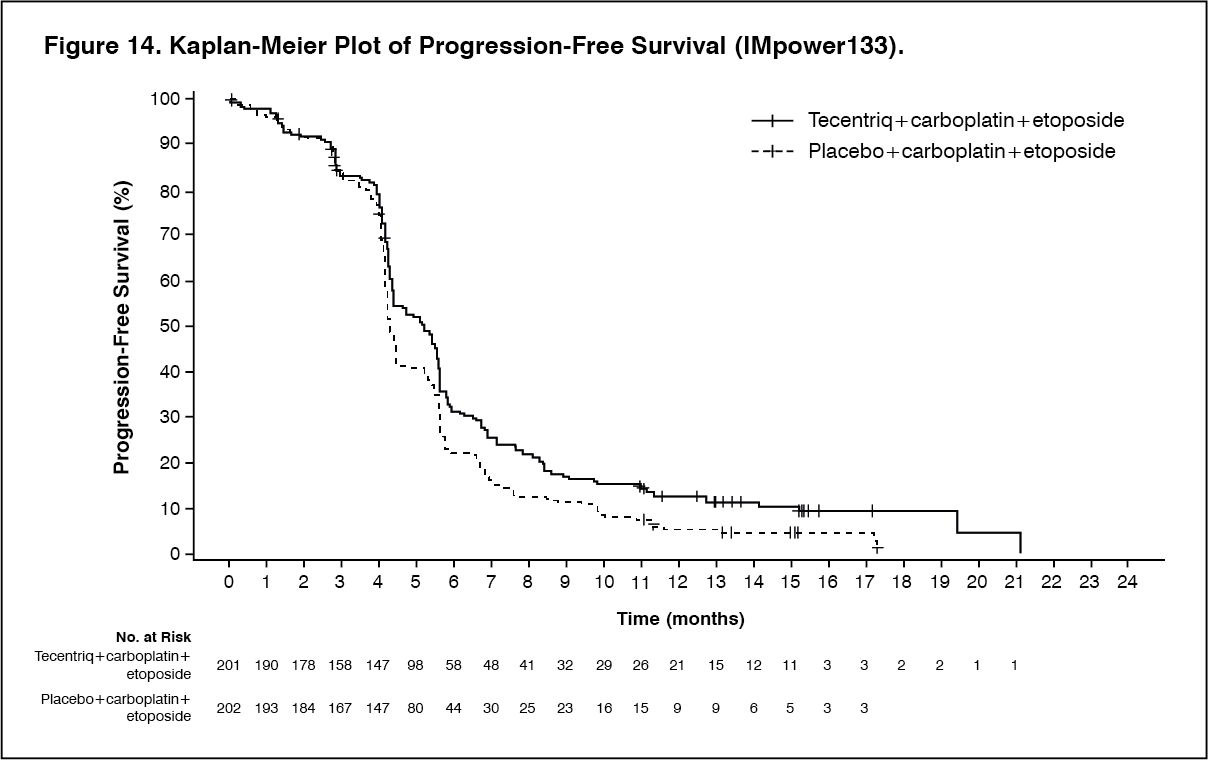 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThis study also included an exploratory analysis of average score changes from baseline in patient-reported symptoms, physical function, and health-related quality of life (measured using the EORTC QLC-C30 and QLC-LC13). On average, patients who received Tecentriq with carboplatin and etoposide reported early and notable improvements in lung cancer-related symptoms (e.g., coughing, chest pain, dyspnea) and physical function. Changes in treatment-related symptoms (e.g., diarrhea, nausea and vomiting, sore mouth, peripheral neuropathy) were comparable between arms throughout induction and most visits through week 54. Overall, patients treated with Tecentriq, carboplatin and etoposide achieved more pronounced and enduring improvements in health-related quality of life (≥10-point score increases at most visits through Week 48) compared to patients treated with placebo, carboplatin and etoposide, who reported nominal improvements (<10-point score increases) at most study treatment visits.
2L NSCLC: OAK: A phase III, open-label, multi-center, international, randomized study, GO28915 (OAK), was conducted to evaluate the efficacy and safety of Tecentriq compared with docetaxel in patients with locally advanced or metastatic NSCLC who have progressed during or following a platinum-containing regimen. A total of 1225 patients were enrolled, with the primary analysis population consisting of the first 850 randomized patients. Eligible patients were stratified by PD-L1 expression status in tumor-infiltrating immune cells (IC), by the number of prior chemotherapy regimens, and by histology. Patients were randomized (1:1) to receive either Tecentriq or docetaxel. This study excluded patients who had a history of autoimmune disease, active or corticosteroid-dependent brain metastases, administration of a live, attenuated vaccine within 28 days prior to enrollment, administration of systemic immunostimulatory agents within 4 weeks or systemic immunosuppressive medications within 2 weeks prior to enrollment. Tumor assessments were conducted every 6 weeks for the first 36 weeks, and every 9 weeks thereafter. Tumor specimens were evaluated prospectively for PD-L1 expression on tumor cells (TC) and IC and the results were used to define the PD-L1 expression subgroups for the analyses described as follows.
The demographic and baseline disease characteristics of the primary analysis population were well balanced between the treatment arms. The median age was 64 years (range: 33 to 85), and 61% of patients were male. The majority of patients were white (70%). Approximately three-fourths of patients had non-squamous disease (74%), 10% had known EGFR mutation, 0.2% had known ALK rearrangements, 10% had CNS metastases at baseline, and most patients were current or previous smokers (82%). Baseline ECOG performance status was 0 (37%) or 1 (63%). Seventy-five percent of patients received only one prior platinum-based therapeutic regimen.
Tecentriq was administered as a fixed dose of 1200 mg by IV infusion every 3 weeks. No dose reduction was allowed. Patients were treated until loss of clinical benefit as assessed by the investigator. Docetaxel was administered 75 mg/m2 by IV infusion on day 1 of each 21 day cycle until disease progression. For all treated patients, the median duration of treatment was 2.1 months for the docetaxel arm and 3.4 months for the Tecentriq arm.
The primary efficacy endpoint was OS. The key results of this study with a median survival follow-up of 21 months are summarized in Table 13. Kaplan-Meier curves for OS in the ITT population are presented in Figure 15. Figure 16 summarizes the results of OS in the ITT and PD-L1 subgroups, demonstrating OS benefit with Tecentriq in all subgroups, including those with PD-L1 expression < 1% in TC and IC. (See Table 13, Figures 15 and 16.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image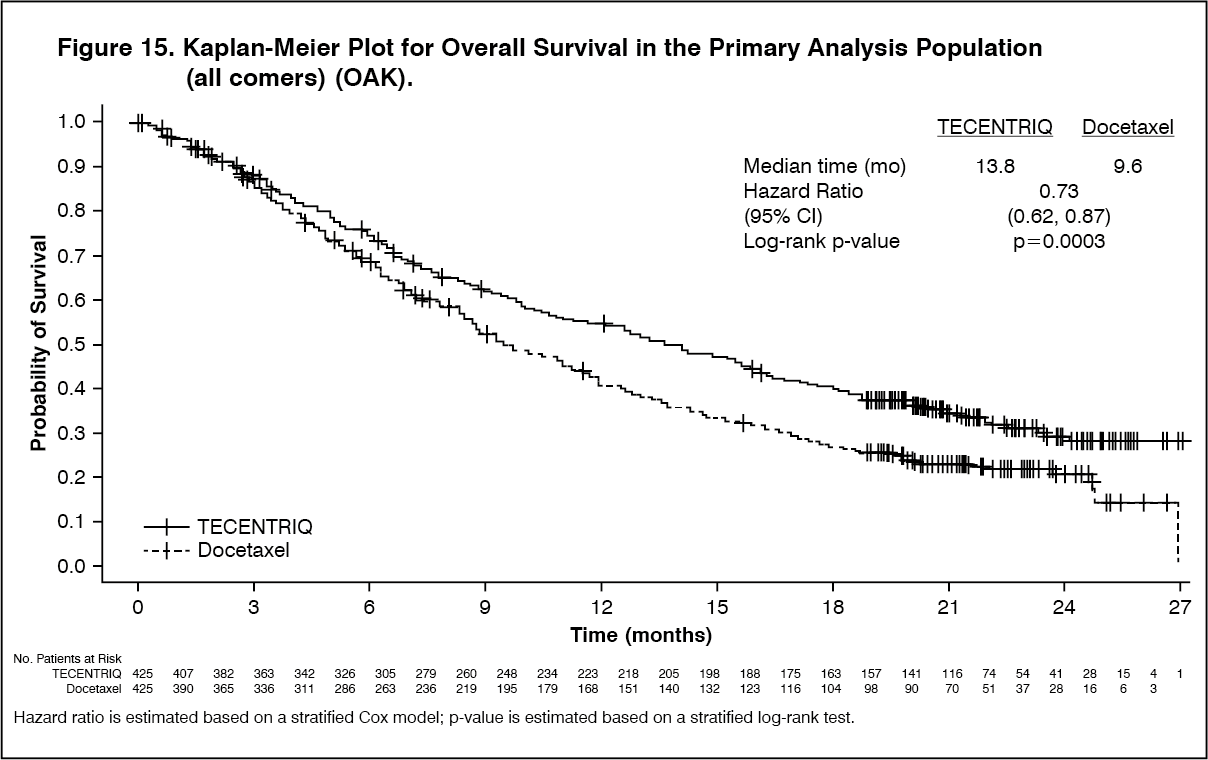 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image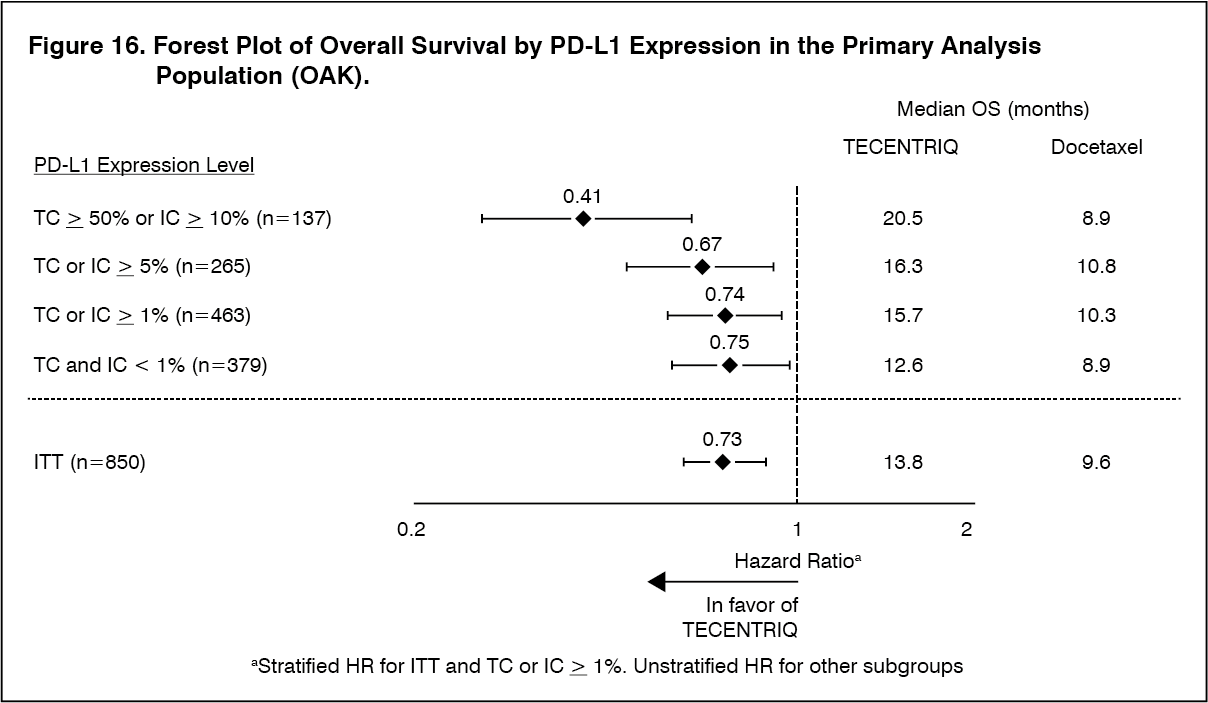 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAn improvement in OS was observed with Tecentriq compared to docetaxel in both non-squamous NSCLC patients (hazard ratio [HR] of 0.73, 95% CI: 0.60, 0.89; median OS of 15.6 vs. 11.2 months for Tecentriq and docetaxel, respectively) and squamous NSCLC patients (HR of 0.73, 95% CI: 0.54, 0.98; median OS of 8.9 vs. 7.7 months for Tecentriq and docetaxel, respectively). The observed OS improvement was consistently demonstrated across subgroups of patients including those with brain metastases at baseline (HR of 0.54, 95% CI: 0.31, 0.94; median OS of 20.1 vs. 11.9 months for Tecentriq and docetaxel respectively) and patients who were never smokers (HR of 0.71, 95% CI: 0.47, 1.08; median OS of 16.3 vs. 12.6 months for Tecentriq and docetaxel, respectively). However, patients with EGFR mutations did not show improved OS with Tecentriq compared to docetaxel (HR of 1.24, 95% CI: 0.71, 2.18; median OS of 10.5 vs. 16.2 months for Tecentriq and docetaxel respectively).
Prolonged time to deterioration of patient-reported pain in chest as measured by the EORTC QLQ-LC13 was observed with Tecentriq compared with docetaxel (HR 0.71, 95% CI: 0.49, 1.05; median not reached in either arm). The time to deterioration in other lung cancer symptoms (i.e. cough, dyspnea, and arm/shoulder pain) as measured by the EORTC QLQ-LC13 was similar between Tecentriq and docetaxel. The average global health status and functioning scores (i.e. physical, role, social, emotional, and cognitive) as measured by the EORTC QLQ-C30 did not show clinically meaningful deterioration over time for both treatment groups, suggesting maintained health-related quality of life and patient-reported functioning for patients remaining on treatment.
POPLAR: A phase II, multi-center, international, randomized, open-label, controlled study GO28753 (POPLAR), was conducted in patients with locally advanced or metastatic NSCLC. The primary efficacy outcome was overall survival. A total of 287 patients were randomized 1:1 to receive either Tecentriq or docetaxel. Randomization was stratified by PD-L1 expression status in IC, by the number of prior chemotherapy regimens and by histology. An updated analysis with a total of 200 deaths observed and a median survival follow-up of 22 months showed a median OS of 12.6 months in patients treated with Tecentriq vs. 9.7 months in patients treated with docetaxel (HR of 0.69, 95% CI: 0.52, 0.92). ORR was 15.3% vs. 14.7% and median DOR was 18.6 months vs. 7.2 months for Tecentriq vs. docetaxel, respectively.
1L TNBC: IMpassion130: A phase III, double-blind, two-arm, randomized, placebo-controlled study, WO29522 (IMpassion130), was conducted to evaluate the efficacy and safety of Tecentriq in combination with nab-paclitaxel, in patients with unresectable locally advanced or metastatic TNBC who had not received prior chemotherapy for metastatic disease. A total of 902 patients were enrolled and stratified by presence of liver metastases, prior taxane treatment, and by PD-L1 expression status in tumor-infiltrating immune cells (IC) (PD-L1 stained tumour-infiltrating immune cells [IC] in <1% of the tumour area vs. ≥1% of the tumour area). Patients were randomized to receive Tecentriq (840 mg) or placebo IV infusions on Days 1 and 15 of every 28-day cycle, plus nab-paclitaxel (100 mg/m2) administered via IV infusion on Days 1, 8 and 15 of every 28-day cycle. Patients received treatment until radiographic disease progression per RECIST v1.1, or unacceptable toxicity. Treatment with Tecentriq could be continued when nab-paclitaxel was stopped due to unacceptable toxicity.
Patients were excluded if they had a history of autoimmune disease; administration of a live, attenuated vaccine within 4 weeks prior to randomization; administration of systemic immunostimulatory agents within 4 weeks or systemic immunosuppressive medications within 2 weeks prior to randomization; untreated or corticosteroid-dependent brain metastases. Tumor assessments were performed every 8 weeks (± 1 week) for the first 12 months after Cycle 1, day 1 and every 12 weeks (± 1 week) thereafter.
The demographic and baseline disease characteristics of the study population were well balanced between the treatment arms. Most patients were women (99.6%). Sixty-seven percent of patients were white (67.5%), 17.8% were Asian, 6.5% were Black or African American, and 4.4% were American Indian or Alaskan Native. The median age was 55 years (range: 20-86). Baseline ECOG performance status was 0 (58.4%) or 1 (41.3%). Overall, 41% of enrolled patients had PD-L1 expression ≥1%, 27% had liver metastases and 7% brain metastases at baseline. Approximately half the patients had received a taxane (51%) or anthracycline (54%) in the (neo)adjuvant setting. Patient demographics and baseline tumor disease in the PD-L1 expression ≥1% population were generally representative of the broader study population.
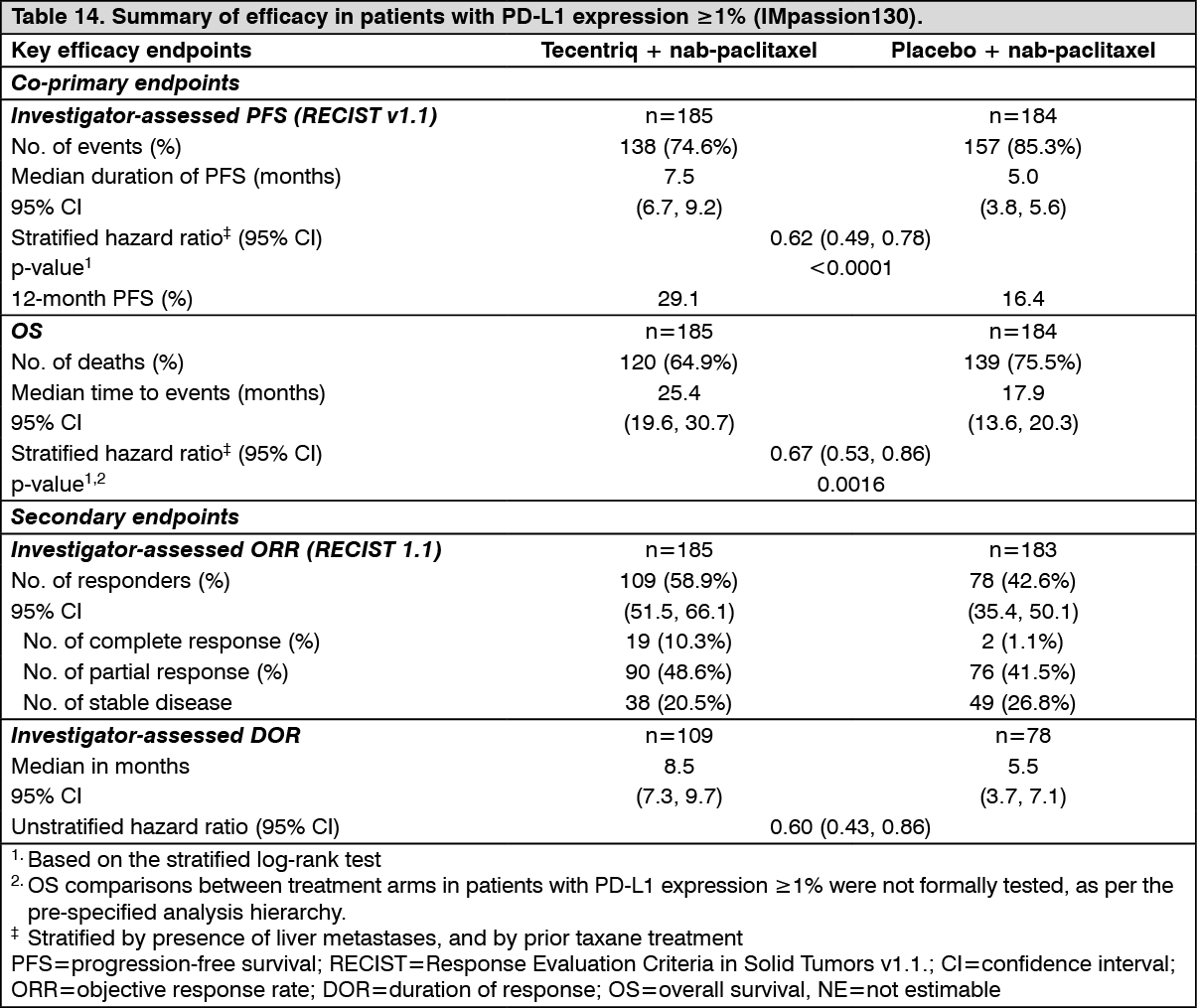
PFS, ORR and DOR results for patients with PD-L1 expression ≥1% with a median survival follow up of 13 months are summarized in Table 14 and Figure 17. In addition, PFS benefit was observed in subgroups.
A final OS analysis was performed in patients with PD-L1 expression ≥1% with a median follow-up of 19.2 months. OS results are presented in Table 14 and Figure 18. (See Table 14, Figures 17 and 18.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image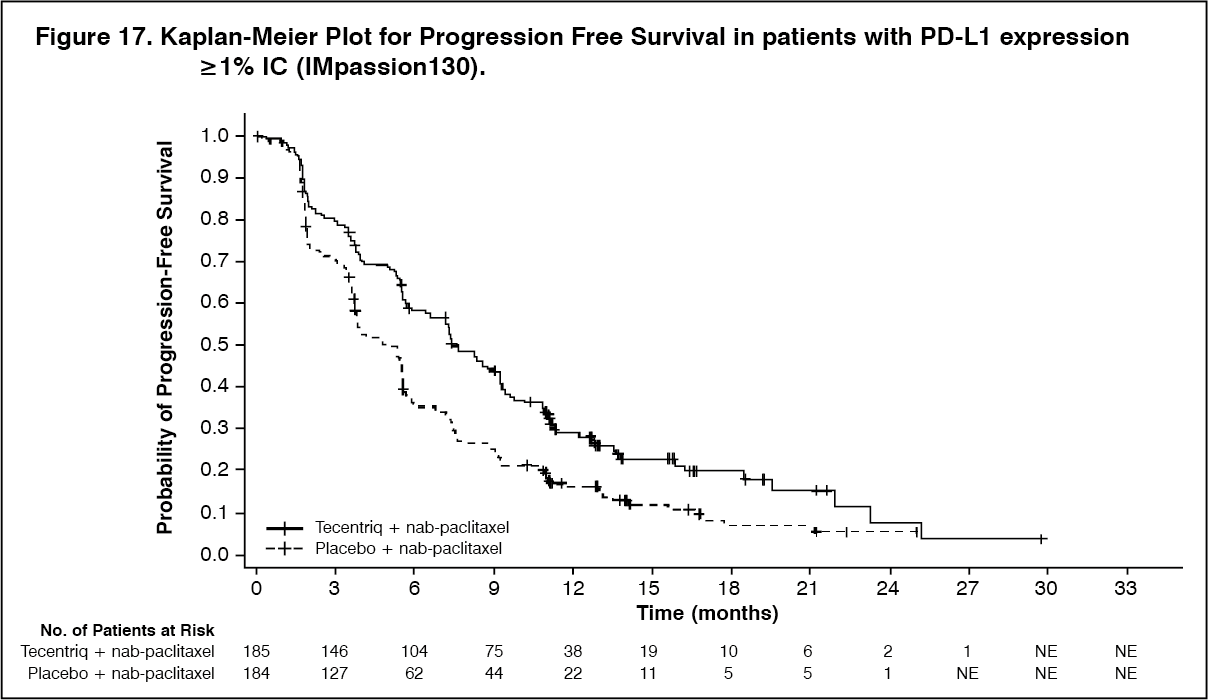 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image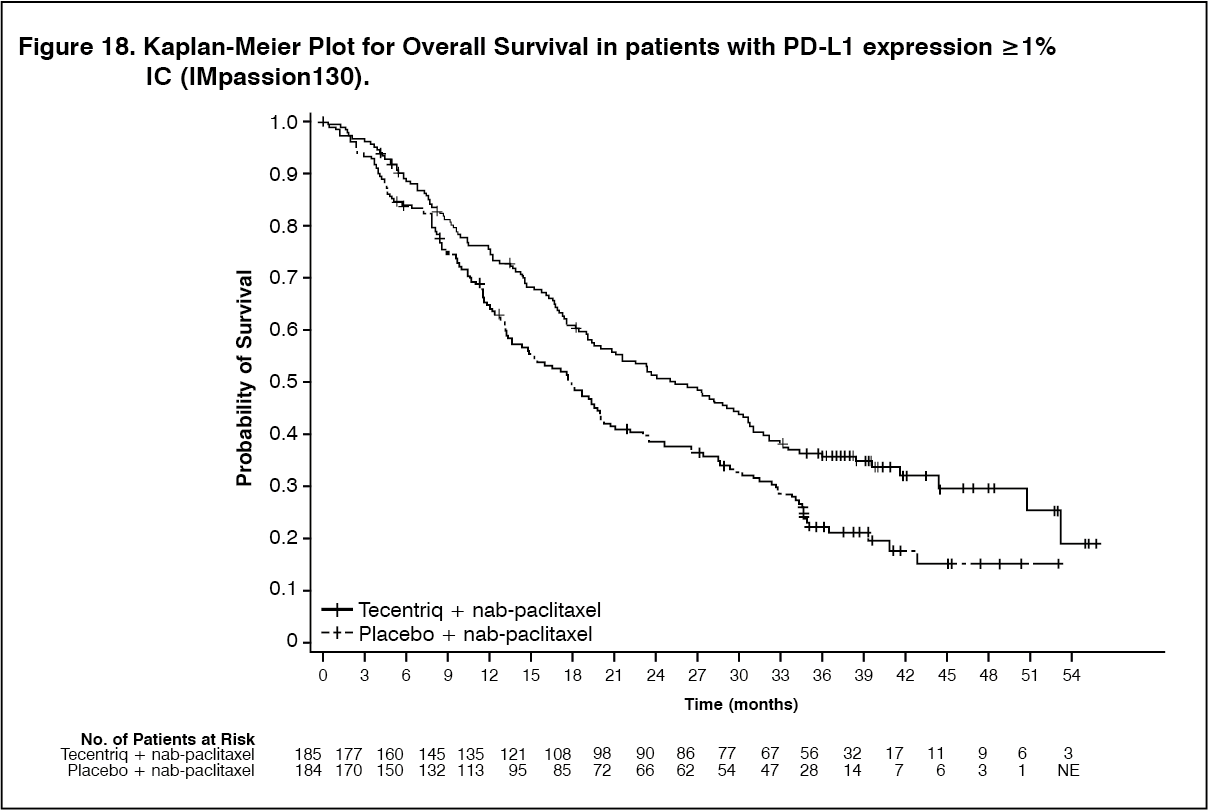 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePatient-reported endpoints measured by the EORTC QLQ-C30 suggest that patients maintained their global health status/health-related quality of life (HRQoL), physical functioning, and role functioning while on treatment. No differences in the time to a ≥10-point deterioration in HRQoL (HR: 0.94; 95% CI: 0.69, 1.28), physical function (HR: 1.02; 95% CI: 0.76, 1.37), or role function (HR: 0.77; 95% CI: 0.57, 1.04) were observed between the two arms. Mean scores at baseline for HRQoL (67.5 Tecentriq and nab-paclitaxel vs. 65.0 placebo and nab-paclitaxel), physical function (82.7 vs. 79.4), and role function (73.6 vs. 71.7) were comparable between arms; as well as throughout the course of treatment. In both arms, HRQoL, physical function and role function remained stable during treatment, with no clinically meaningful changes (a ≥10-point difference from baseline mean score) observed.
HCC: IMbrave150: A global phase III, randomized, multi-center, open-label study, YO40245 (IMbrave150), was conducted to evaluate the efficacy and safety of Tecentriq in combination with bevacizumab, in patients with locally advanced or metastatic and/or unresectable HCC, who have not received prior systemic treatment. A total of 501 patients were randomized (2:1) to receive either Tecentriq 1200 mg and 15 mg/kg of bevacizumab every 3 weeks administered via IV infusion, or sorafenib 400 mg orally twice per day. Randomization was stratified by geographic region (Asia excluding Japan vs. rest of world), macrovascular invasion and/or extrahepatic spread (presence vs. absence), baseline AFP (<400 vs. ≥400 ng/mL) and ECOG performance status (0 vs. 1). Patients in both arms received treatment until loss of clinical benefit, or unacceptable toxicity. Patients could discontinue either Tecentriq or bevacizumab (e.g., due to adverse events) and continue on single-agent therapy until loss of clinical benefit or unacceptable toxicity associated with the single-agent.
The study enrolled adults who were Child-Pugh A, ECOG 0/1 and who had not received prior systemic treatment. Bleeding (including fatal events) is a known adverse reaction with bevacizumab and upper gastrointestinal bleeding is a common and life threatening complication in patients with HCC. Hence, patients were required to be evaluated for the presence of varices within 6 months prior to treatment, and were excluded if they had variceal bleeding within 6 months prior to treatment, untreated or incompletely treated varices with bleeding or high risk of bleeding. Patients were also excluded if they had moderate or severe ascites; history of hepatic encephalopathy; a history of autoimmune disease; administration of a live, attenuated vaccine within 4 weeks prior to randomization; administration of systemic immunostimulatory agents within 4 weeks or systemic immunosuppressive medications within 2 weeks prior to randomization; untreated or corticosteroid-dependent brain metastases. Tumor assessments were performed every 6 weeks for the first 54 weeks following Cycle 1, Day 1, then every 9 weeks thereafter.
The demographic and baseline disease characteristics of the study population were well balanced between the treatment arms. The median age was 65 years (range: 26 to 88 years) and 83% were male. The majority of patients were Asian (57%) and white (35%). 40% were from Asia (excluding Japan), while 60% were from rest of world. Approximately 75% of patients presented with macrovascular invasion and/or extrahepatic spread and 37% had a baseline AFP ≥400 ng/mL. Baseline ECOG performance status was 0 (62%) or 1 (38%). The primary risk factors for the development of HCC were Hepatitis B virus infection in 48% of patients, Hepatitis C virus infection in 22% of patients, and non-viral disease in 31% of patients. HCC was categorized as Barcelona Clinic Liver Cancer (BCLC) stage C in 82% of patients, stage B in 16% of patients, and stage A in 3% of patients.
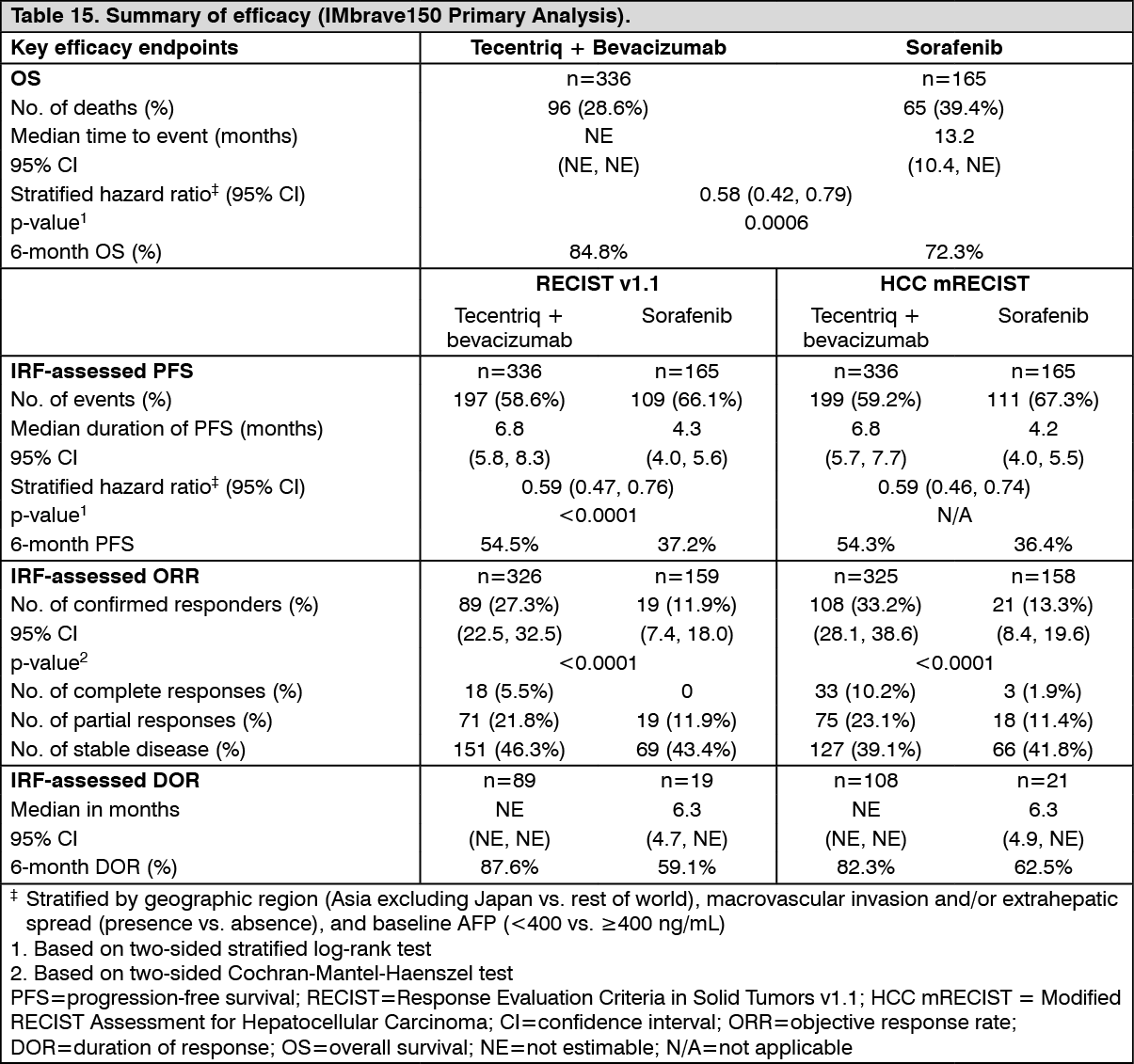
The co-primary efficacy endpoints were OS and IRF-assessed PFS according to RECIST v1.1. At the time of the primary analysis, patients had a median survival follow up time of 8.6 months. The data demonstrated a statistically significant improvement in OS and PFS as assessed by IRF per RECIST v1.1 with Tecentriq + bevacizumab compared to sorafenib. A statistically significant improvement was also observed in confirmed objective response rate (ORR) by IRF per RECIST v1.1 and HCC modified RECIST (mRECIST). The key efficacy results from the primary analysis are summarized in Table 15.
A descriptive updated efficacy analysis was performed with a median survival follow up time of 15.6 months. The key results from the updated analysis are summarized in Table 16. Kaplan-Meier curves for OS (updated analysis) and PFS (primary analysis) are presented in Figures 19 and 20, respectively. (See Tables 15 and 16, and Figures 19 and 20.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image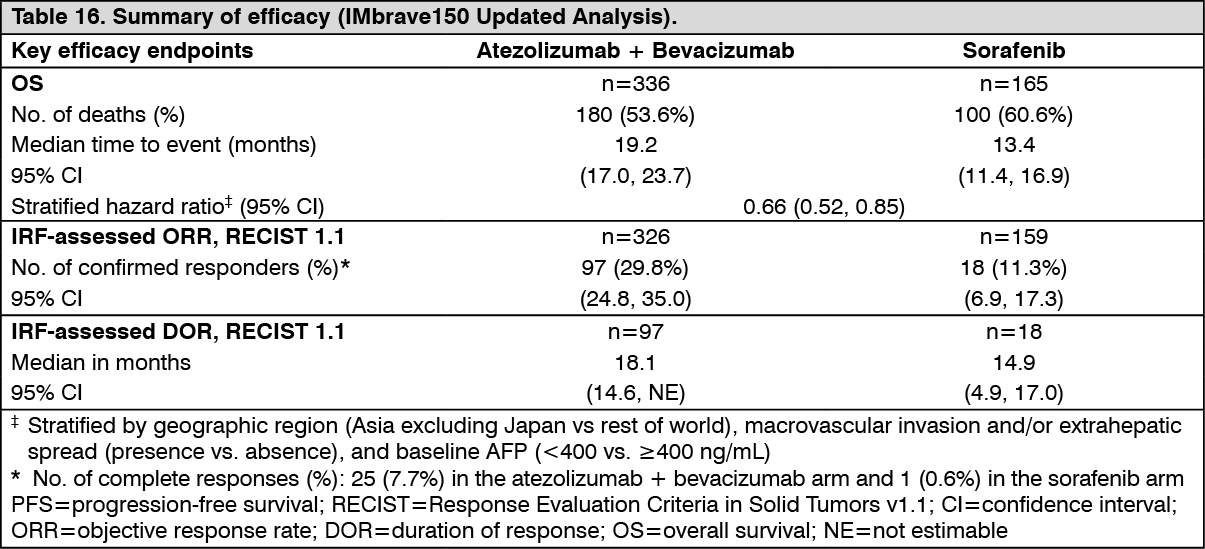 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image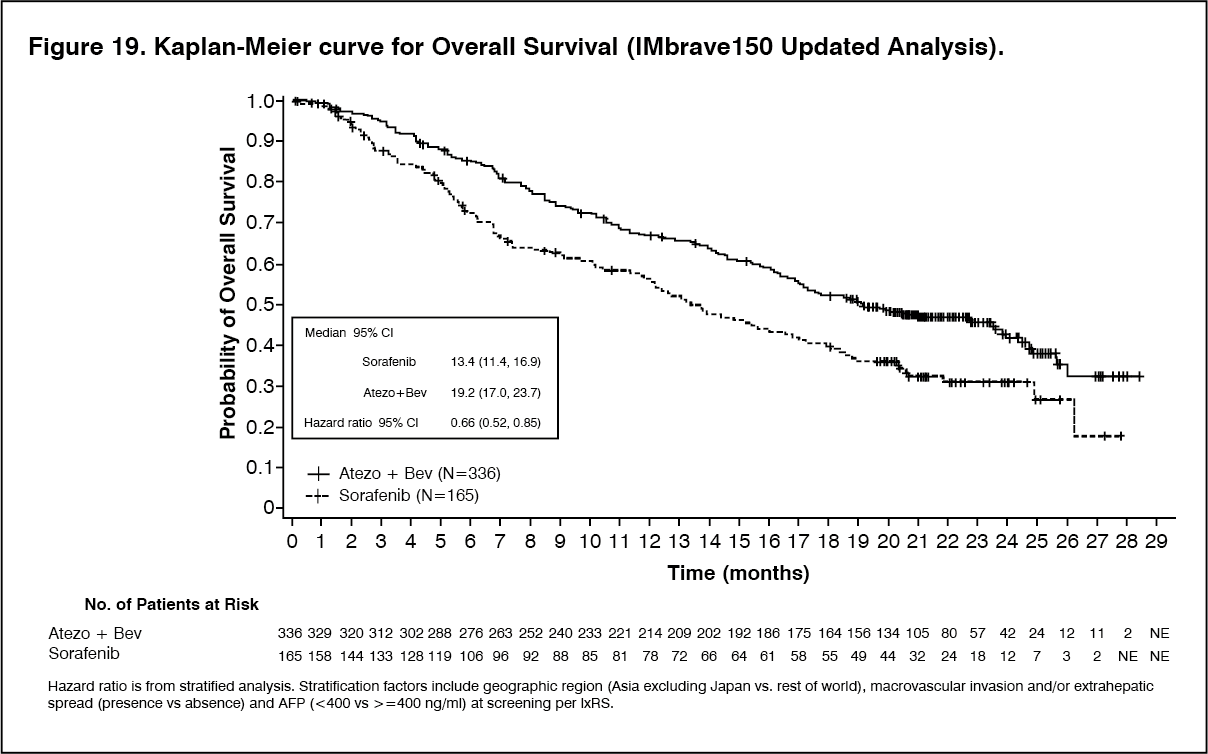 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image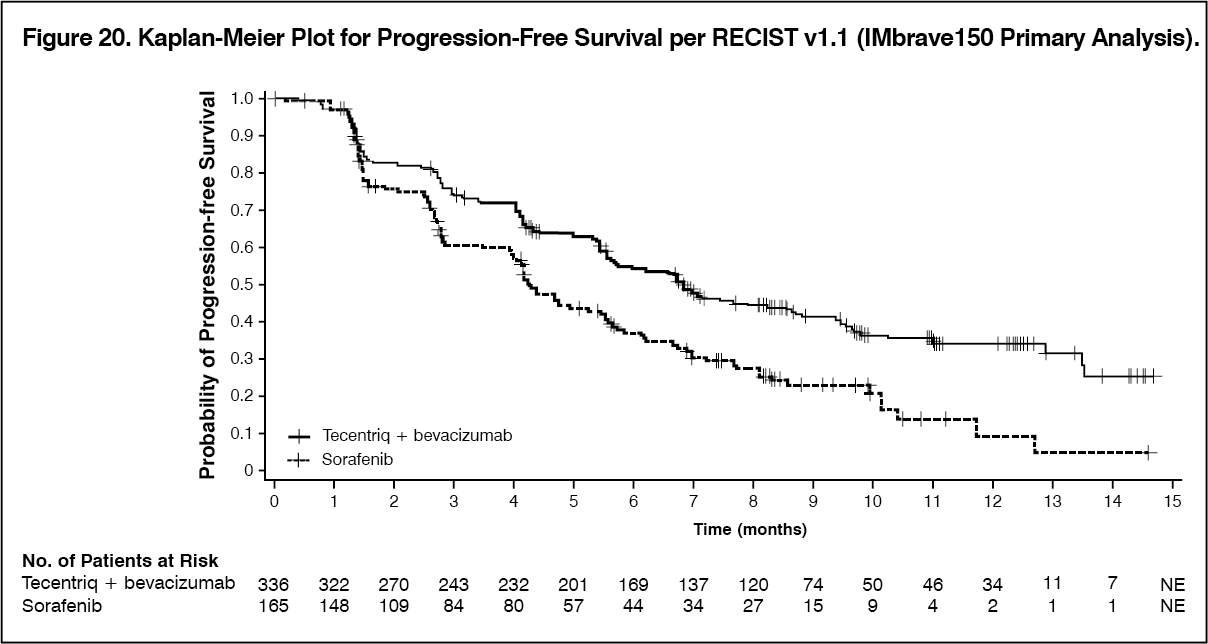 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe study evaluated patient-reported outcomes using the EORTC QLQ-C30 and EORTC QLQ-HCC18 questionnaires. Time to deterioration (TTD) of patient-reported physical functioning, role functioning, and global health status/quality of life (GHS/QoL) on the EORTC QLQ-C30 were pre-specified secondary endpoints. TTD was defined as the time from randomization to the first deterioration (decrease from baseline of ≥10 points) maintained for two consecutive assessments, or one assessment followed by death from any cause within 3 weeks. Compared with sorafenib, treatment with Tecentriq and bevacizumab delayed deterioration of patient-reported physical functioning (median TTD: 13.1 vs. 4.9 months; HR 0.53, 95% CI 0.39, 0.73), role functioning (median TTD: 9.1 vs. 3.6 months; HR 0.62, 95% CI 0.46, 0.84), and GHS/QoL (median TTD: 11.2 vs. 3.6 months; HR 0.63, 95% CI 0.46, 0.85). In pre-specified exploratory analyses, compared with sorafenib, treatment with Tecentriq and bevacizumab also delayed deterioration of patient-reported symptoms (i.e. appetite loss, diarrhea, fatigue, pain, and jaundice) on the EORTC QLQ-C30 and EORTC QLQ-HCC18.
GO30140: A global, open-label, multi-center, multi-arm Phase Ib study (GO30140) was also conducted in patients with solid tumors. Arm F of the study used a randomized design to evaluate the safety and efficacy of Tecentriq administered in combination with bevacizumab versus Tecentriq monotherapy in patients with advanced or metastatic and/or unresectable HCC who had not received prior systemic treatment. The primary efficacy endpoint was PFS assessed by IRF according to RECIST v1.1. A total of 119 patients were randomized 1:1 to receive either Tecentriq (1200 mg) and bevacizumab (15 mg/kg) by IV infusion every 3 weeks or Tecentriq (1200 mg) every 3 weeks. At the time of the primary analysis, the median survival follow up was 6.6 months. The combination of Tecentriq with bevacizumab showed statistically significant PFS benefit compared to Tecentriq monotherapy (HR of 0.55, 80% CI: 0.40, 0.74, p-value = 0.0108) with a median PFS of 5.6 months in patients treated with Tecentriq and bevacizumab, vs 3.4 months in patients treated with Tecentriq monotherapy.
Immunogenicity: As with all therapeutic proteins, there is the potential for immune response to atezolizumab. Across multiple phase III studies, 13.1% to 36.4% of patients developed treatment-emergent anti-drug antibodies (ADAs) and 4.3% to 19.7% of patients developed neutralling antibodies (Nabs). ADA and Nab status appeared to have no clinically relevant impact on atezolizumab pharmacokinetics, efficacy or safety.
Immunogenicity assay results are highly dependent on several factors including assay sensitivity and specificity, assay methodology, sample handling, timing of sample collection, concomitant medications and underlying disease. For these reasons, comparison of incidence of antibodies to Tecentriq with the incidence of antibodies to other products may be misleading.
Pharmacokinetics: The pharmacokinetics of atezolizumab have been characterized in patients in multiple clinical trials at doses of 0.01 mg/kg to 20 mg/kg and 1200 mg every 3 weeks, as well as 840 mg every 2 weeks. Exposure to atezolizumab increased dose proportionally over the dose range of 1 mg/kg to 20 mg/kg. A population analysis that included 472 patients described atezolizumab pharmacokinetics for the dose range of 1-20 mg/kg with a linear two-compartment disposition model with first-order elimination. Based on pharmacokinetic modelling, the overall exposure of atezolizumab administered at dose of 840 mg every 2 weeks, 1200 mg every 3 weeks and 1680 mg every 4 weeks are comparable. A population pharmacokinetic analysis suggests that steady state is obtained after 6 to 9 weeks after multiple dose. The maximum systemic accumulation ratio across dosing regimen is 3.3.
Based on an analysis of exposure, safety and efficacy data, the following factors have no clinically relevant effect: age (2-89 years), body weight, gender, positive ADA status, albumin levels, tumor burden, region or ethnicity, renal impairment, mild hepatic impairment, level of PD-L1 expression, or ECOG status.
Absorption: Tecentriq is administered as an IV infusion. There have been no studies performed with other routes of administration.
Distribution: A population pharmacokinetic analysis indicates that central compartment volume of distribution (V1) is 3.28 L and volume at steady state (Vss) is 6.91 L in the typical patient.
Metabolism: The metabolism of Tecentriq has not been directly studied. Antibodies are cleared principally by catabolism.
Elimination: A population pharmacokinetic analysis indicates that the clearance of atezolizumab is 0.200 L/day and the typical terminal elimination half-life (t½) is 27 days.
Pharmacokinetics in Special Populations: Pediatric Population: The pharmacokinetic results from one early-phase, multi-centre open-label study that was conducted in pediatric (<18 years, n=69) and young adult patients (18-30 years, n=18), show that the clearance and volume of distribution of atezolizumab were comparable between pediatric patients receiving 15 mg/kg and young adult patients receiving 1200 mg of atezolizumab every 3 weeks when normalized by body weight, with exposure trending lower in pediatric patients as body weight decreased. These differences were not associated with a decrease in atezolizumab concentrations below the therapeutic target exposure. Data for children <2 years is limited thus no definitive conclusions can be made.
Geriatric Population: No dedicated studies of Tecentriq have been conducted in geriatric patients. The effect of age on the pharmacokinetics of atezolizumab was assessed in a population pharmacokinetic analysis. Age was not identified as a significant covariate influencing atezolizumab pharmacokinetics based on patients of age range of 21-89 years (n=472), and median of 62 years of age. No clinically important difference was observed in the pharmacokinetics of atezolizumab among patients <65 years (n=274), patients between 65-75 years (n=152) and patients >75 years (n=46) (see Special Dosage Instructions under Dosage & Administration).
Renal impairment: No dedicated studies of Tecentriq have been conducted in patients with renal impairment. In the population pharmacokinetic analysis, no clinically important differences in the clearance of atezolizumab were found in patients with mild (eGFR 60 to 89 ml/min/1.73 m2; n=208) or moderate (eGFR 30 to 59 ml/min/1.73 m2; n=116) renal impairment compared to patients with normal (eGFR greater than or equal to 90 ml/min/1.73 m2; n=140) renal function. Only a few patients had severe renal impairment (eGFR 15 to 29 ml/min/1.73 m2; n=8) (see Special Dosage Instructions under Dosage & Administration).
Hepatic impairment: No dedicated studies of Tecentriq have been conducted in patients with hepatic impairment. In the population pharmacokinetic analysis, there were no clinically important differences in the clearance of atezolizumab between patients with mild hepatic impairment (bilirubin ≤ ULN and AST > ULN or bilirubin > 1.0 to 1.5 x ULN and any AST) or moderate hepatic impairment (bilirubin >1.5 to 3 x ULN and any AST). No data are available in patients with severe (bilirubin >3.0 x ULN and any AST) hepatic impairment. Hepatic impairment was defined by the National Cancer Institute (NCI) criteria of hepatic dysfunction (see Special Dosage Instructions under Dosage & Administration).
Toxicology: Nonclinical Safety: Carcinogenicity: No carcinogenicity studies have been conducted with Tecentriq.
Genotoxicity: No mutagenicity studies have been conducted with Tecentriq.
Impairment of Fertility: No fertility studies have been conducted with Tecentriq; however, assessment of the cynomolgus monkey male and female reproductive organs was included in the chronic toxicity study. Tecentriq had an effect on menstrual cycles in all female monkeys in the 50 mg/kg dose group characterized by an irregular cycle pattern during the dosing phase and correlated with the lack of fresh corpora lutea in the ovaries at the terminal necropsy; this effect was reversible during the dose-free recovery period. There was no effect on the male reproductive organs.
Reproductive Toxicity: No reproductive or teratogenicity studies in animals have been conducted with Tecentriq. The PD-L1/PD-1 signaling pathway is well established as essential in maternal/fetal tolerance and embryo-fetal survival during gestation. Administration of Tecentriq is expected to have an adverse effect on pregnancy and poses a risk to the human fetus, including embryo lethality.
Other: Not applicable.