Summary of the safety profile: In placebo-controlled trials in 1,617 MS patients treated with natalizumab for up to 2 years (placebo: 1,135), adverse events leading to discontinuation of therapy occurred in 5.8% of patients treated with natalizumab (placebo: 4.8%). Over the 2-year duration of the studies, 43.5% of patients treated with natalizumab reported adverse reactions (placebo: 39.6%).
The highest incidence of adverse reactions identified from placebo-controlled trials in multiple sclerosis patients with natalizumab given at the recommended dose, are reported as dizziness, nausea, urticaria and rigors associated with infusions.
Tabulated list of adverse reactions: Adverse reactions reported with natalizumab with an incidence of 0.5% greater than reported with placebo are shown as follows.
The reactions are reported as MedDRA preferred terms under the MedDRA primary system organ class. Frequencies were defined as follows: Common (≥ 1/100 to < 1/10), uncommon (≥1/1,000 to < 1/100).
Within each frequency grouping, adverse reactions are presented in order of decreasing seriousness. (See Table 2.)
 Click on icon to see table/diagram/image
Description of selected adverse reactions: Infusion reactions:
Click on icon to see table/diagram/image
Description of selected adverse reactions: Infusion reactions: In 2-year controlled clinical trials in MS patients, an infusion related event was defined as an adverse event occurring during the infusion or within 1 hour of the completion of the infusion. These occurred in 23.1% of MS patients treated with natalizumab (placebo: 18.7%). Events reported more commonly with natalizumab than with placebo included dizziness, nausea, urticaria and rigors.
Hypersensitivity reactions: In 2-year controlled clinical trials in MS patients, hypersensitivity reactions occurred in up to 4% of patients. Anaphylactic/anaphylactoid reactions occurred in less than 1% of patients receiving TYSABRI. Hypersensitivity reactions usually occurred during the infusion or within the 1 hour period after the completion of the infusion (See Precautions). In post-marketing experience, there have been reports of hypersensitivity reactions which have occurred with one or more of the following associated symptoms: hypotension, hypertension, chest pain, chest discomfort, dyspnoea, angioedema, in addition to more usual symptoms such as rash and urticaria.
Immunogenicity: In 10% of patients antibodies against natalizumab were detected in 2-year controlled clinical trials in MS patients. Persistent anti-natalizumab antibodies (one positive test reproducible on retesting at least 6 weeks later) developed in approximately 6% of patients. Antibodies were detected on only one occasion in an additional 4% of patients. Persistent antibodies were associated with a substantial decrease in the effectiveness of TYSABRI and an increased incidence of hypersensitivity reactions. Additional infusion-related reactions associated with persistent antibodies included rigors, nausea, vomiting and flushing (see Precautions).
If, after approximately 6 months of therapy, persistent antibodies are suspected, either due to reduced efficacy or due to occurrence of infusion-related events, they may be detected and confirmed with a subsequent test 6 weeks after the first positive test. Given that efficacy may be reduced or the incidence of hypersensitivity or infusion-related reactions may be increased in a patient with persistent antibodies, treatment should be discontinued in patients who develop persistent antibodies.
Infections, including PML and opportunistic infections: In 2-year controlled clinical trials in MS patients, the rate of infection was approximately 1.5 per patient year in both natalizumab- and placebo-treated patients. The nature of the infections was generally similar in natalizumab- and placebo-treated patients. A case of
cryptosporidium diarrhoea was reported in MS clinical trials. In other clinical trials, cases of additional opportunistic infections have been reported, some of which were fatal. The majority of patients did not interrupt natalizumab therapy during infections and recovery occurred with appropriate treatment.
In clinical trials, herpes infections (Varicella-Zoster virus, Herpes-simplex virus) occurred slightly more frequently in natalizumab-treated patients than in placebo-treated patients. In post marketing experience, serious, life-threatening, and sometimes fatal cases of encephalitis and meningitis caused by herpes simplex or varicella zoster have been reported in multiple sclerosis patients receiving TYSABRI. The duration of treatment with TYSABRI prior to onset ranged from a few months to several years (see Precautions).
In postmarketing experience, rare cases of ARN have been observed in patients receiving TYSABRI. Some cases have occurred in patients with central nervous system (CNS) herpes infections (e.g. herpes meningitis and encephalitis). Serious cases of ARN, either affecting one or both eyes, led to blindness in some patients. The treatment reported in these cases included anti-viral therapy and in some cases, surgery (see Precautions).
Cases of PML have been reported from clinical trials, post-marketing observational studies and post-marketing passive surveillance. PML usually leads to severe disability or death (see Precautions).
Cases of JCV GCN have also been reported during postmarketing use of TYSABRI. Symptoms of JCV GCN are similar to PML.
Hepatic Events: Spontaneous cases of serious liver injuries, increased liver enzymes, hyperbilirubinaemia have been reported during the post marketing phase (see Precautions).
Anaemia and haemolytic anaemia: Rare, serious cases of anaemia and haemolytic anaemia have been reported in patients treated with TYSABRI in post-marketing observational studies.
Malignancies: No differences in incidence rates or the nature of malignancies between natalizumab- and placebo treated patients were observed over 2 years of treatment. However, observation over longer treatment periods is required before any effect of natalizumab on malignancies can be excluded. See Contraindications.
Effects on laboratory tests: In 2-year controlled clinical trials in MS patients TYSABRI treatment was associated with increases in circulating lymphocytes, monocytes, eosinophils, basophils and nucleated red blood cells. Elevations in neutrophils were not seen. Increases from baseline for lymphocytes, monocytes, eosinophils and basophils ranged from 35% to 140% for individual cell types but mean cell counts remained within normal ranges. During treatment with TYSABRI, small reductions in haemoglobin (mean decrease 0.6 g/dl), haematocrit (mean decrease 2%) and red blood cell counts (mean decrease 0.1 x 10
6/l) were seen. All changes in haematological variables returned to pre-treatment values, usually within 16 weeks of last dose of the medicinal product and the changes were not associated with clinical symptoms. In post-marketing experience, there have also been reports of eosinophilia (eosinophil count >1,500/mm
3) without clinical symptoms. In such cases where TYSABRI therapy was discontinued the elevated eosinophil levels resolved.
Paediatric population: Serious adverse events were evaluated in 621 MS paediatric patients included in a meta-analysis (see also Pharmacology: Pharmacodynamics under Actions). Within the limitations of these data, there were no new safety signals identified in this patient population. 1 case of herpes meningitis was reported in the meta-analysis. No cases of PML were identified in the meta-analysis, however, PML has been reported in natalizumab treated paediatric patients in the post-marketing setting.
Reporting of suspected adverse reactions: Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image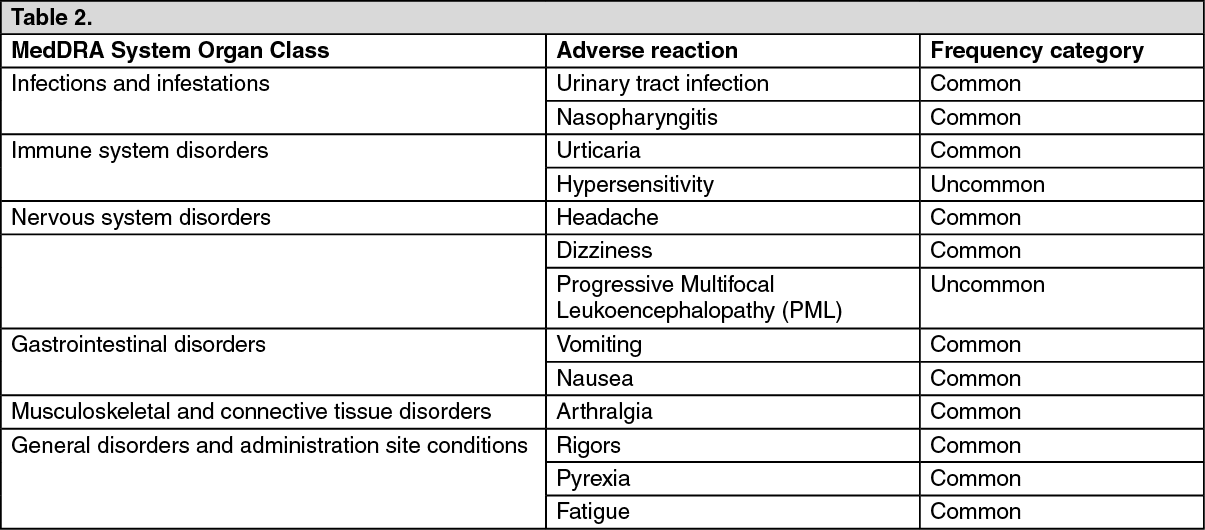 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out