Summary of the Safety Profile: The most serious adverse drug reactions reported in patients receiving Zometa in the approved indications are: Anaphylactic reaction, ocular adverse events, ONJ, atypical femoral fracture, atrial fibrillation, renal function impairment, acute phase reaction and hypocalcemia. The frequencies for each of these adverse reactions are shown as follows or in Adverse Drug Reactions from Spontaneous Reports and Literature Cases.
Frequencies of adverse reactions for Zometa 4 mg are mainly based on data collected from chronic treatment. Adverse reactions to Zometa are usually mild and transient and similar to those reported for other bisphosphonates. Those reactions can be expected to occur in approximately
1/
3 of patients treated with Zometa.
Within 3 days after Zometa administration, an acute phase reaction has commonly been reported, with symptoms including pyrexia, fatigue, bone pain, chills and influenza-like illness, arthritis with subsequent joint swelling; these symptoms usually resolve within a few days (see Description of Selected Adverse Reactions). Cases of arthralgia and myalgia have commonly been reported.
Very commonly, the reduction in renal calcium excretion is accompanied by a fall in serum phosphate levels, which is asymptomatic not requiring treatment. The serum calcium may fall to asymptomatic hypocalcemic levels.
Gastrointestinal reactions eg, nausea and vomiting have been commonly reported following IV infusion of Zometa. Uncommonly local reactions at the infusion site eg, redness or swelling and/or pain were also observed.
Anorexia was commonly reported in patients treated with Zometa 4 mg.
Rash or pruritus has been uncommonly observed. As with other bisphosphonates, cases of conjunctivitis have been reported.
Based on pooled analysis of placebo-controlled studies, severe anemia (Hb <8 g/dL) was commonly reported in patients receiving Zometa 4 mg.
Adverse drug reactions from clinical trials are listed according to system organ classes in MedDRA. Within each system organ class, the adverse drug reactions are ranked by frequency, with the most frequent reactions first. Within each frequency grouping, adverse drug reactions are presented in order of decreasing seriousness. In addition, the corresponding frequency category using the following convention (CIOMS III) is also provided for each adverse drug reaction: Very common (≥1/10), common (≥1/100, <1/10), uncommon (≥1/1000, <1/100), rare (≥1/10,000, <1/1000), very rare (<1/10,000), including isolated reports.
Blood and Lymphatic System Disorders: Common: Anemia. Uncommon: Thrombocytopenia, leucopenia. Rare: Pancytopenia.
Immune System Disorders: Uncommon: Hypersensitivity reaction. Rare: Angioedema.
Nervous System Disorders: Common: Headache, paresthesia. Uncommon: Dizziness, taste disturbance, hypoesthesia, hyperesthesia, tremor.
Psychiatric Disorders: Common: Sleep disorder. Uncommon: Anxiety. Rare: Confusion.
Eye Disorders: Common: Conjunctivitis. Uncommon: Blurred vision.
Gastrointestinal Disorders: Common: Nausea, vomiting, anorexia, constipation. Uncommon: Diarrhea, abdominal pain, dyspepsia, stomatitis, dry mouth.
Respiratory, Thoracic and Mediastinal Disorders: Uncommon: Dyspnea, cough.
Skin and Subcutaneous Tissue Disorders: Common: Hyperhidrosis. Uncommon: Pruritus, rash (including erythematous and macular rash).
Musculoskeletal and Connective Tissue Disorders: Common: Bone pain, myalgia, arthralgia, generalized pain. Uncommon: Osteonecrosis of the jaw (ONJ), muscle cramps.
Cardiovascular Disorders: Rare: Bradycardia.
Vascular Disorders: Common: Hypertension. Uncommon: Hypotension.
Renal and Urinary Disorders: Common: Renal impairment. Uncommon: Acute renal failure, hematuria, proteinuria.
General Disorders and Administration Site Conditions: Common: Acute phase reaction, pyrexia, influenza-like illness (including fatigue, chills, malaise and flushing), peripheral edema, asthenia. Uncommon: Injection site reactions (including pain, irritation, swelling, induration, redness), chest pain, increased weight. Rare: Arthritis and joint swelling as a symptom of acute phase reaction.
Investigations: Very Common: Hypophosphatemia. Common: Increased blood creatinine and blood urea, hypocalcemia. Uncommon: Hypomagnesemia, hypokalemia. Rare: Hyperkalemia, hypernatremia
.
Adverse Drug Reactions from Spontaneous Reports and Literature Cases (Frequency Not Known): The following adverse reactions have been reported during post-marketing experience with Zometa via spontaneous case reports and literature cases. Because these reactions are reported voluntarily from a population of uncertain size and are subject to confounding factors, it is not possible to reliably estimate their frequency (which is therefore categorized as not known) or establish a causal relationship to drug exposure.
Immune System Disorders: Anaphylactic reaction/shock.
Nervous System Disorders: Somnolence.
Eye Disorders: Uveitis, episcleritis, scleritis and orbital inflammation.
Cardiac Disorders: Atrial fibrillation.
Vascular Disorders: Hypotension leading to syncope or circulatory collapse, primarily in patients with underlying risk factors.
Respiratory, Thoracic and Mediastinal Disorders: Bronchospasms, interstitial lung disease (ILD).
Skin and Subcutaneous Tissue Disorders: Urticaria.
Musculoskeletal and Connective Tissue Disorders: Severe and occasionally incapacitating bone, joint, and/or muscle pain, atypical subtrochanteric and diaphyseal femoral fractures (bisphosphonate class adverse reaction, including Zometa).
Description of Selected Adverse Reactions: Renal Function Impairment: Zometa has been associated with reports of renal function impairment. In a pooled analysis of safety data from Zometa registration trials for the prevention of skeletal-related events in patients with advanced malignancy involving bone, the frequency of renal function impairment adverse events suspected to be related to Zometa (adverse reactions) was as follows: Multiple myeloma (3.2%), prostate cancer (3.1%), breast cancer (4.3%), lung and other solid tumors (3.2%). Factors that may increase the potential for deterioration in renal function include dehydration, preexisting renal impairment, multiple cycles of Zometa or other bisphosphonates, as well as concomitant use of nephrotoxic medicinal products or using a shorter infusion time than currently recommended. Renal deterioration, progression to renal failure and dialysis have been reported in patients after the initial dose or a single dose of Zometa (see Precautions).
Osteonecrosis of the Jaw (ONJ): Cases of osteonecrosis (primarily of the jaws) have been reported predominantly in cancer patients treated with bisphosphonates, including Zometa (uncommon). Many of these patients had signs of local infection including osteomyelitis, and the majority of the reports refer to cancer patients following tooth extractions or other dental surgeries. Osteonecrosis of the jaw has multiple well documented risk factors including a diagnosis of cancer, concomitant therapies (eg, chemotherapy, radiotherapy, corticosteroids) and co-morbid conditions (eg, anemia, coagulopathies, infection, preexisting oral disease). Although causality has not been determined, it is prudent to avoid dental surgery as recovery may be prolonged (see Precautions). Data suggests a greater frequency of reports of ONJ based on tumor type (advanced breast cancer, multiple myeloma).
Acute Phase Reaction: This adverse drug reaction consists of a constellation of symptoms that includes pyrexia, fatigue, bone pain, chills, influenza-like illness, arthritis with subsequent joint swelling. The onset time is ≤3 days post-Zometa infusion and the reaction is also referred to using the terms "flu-like" or "post-dose" symptoms; these symptoms usually resolve within a few days.
Atrial Fibrillation: In one 3-year, randomized, double-blind controlled trial that evaluated the efficacy and safety of zoledronic acid 5 mg once yearly versus placebo in the treatment of postmenopausal osteoporosis (PMO), the overall incidence of atrial fibrillation was 2.5% (96 out of 3862) and 1.9% (75 out of 3852) in patients receiving zoledronic acid 5 mg and placebo, respectively. The rate of atrial fibrillation serious adverse events was 1.3% (51 out of 3862) and 0.6% (22 out of 3852) in patients receiving zoledronic acid 5 mg and placebo, respectively. The imbalance observed in this trial has not been observed in other trials with zoledronic acid, including those with Zometa (zoledronic acid) 4 mg every 3-4 weeks in oncology patients. The mechanism behind the increased incidence of atrial fibrillation in this single clinical trial is unknown.


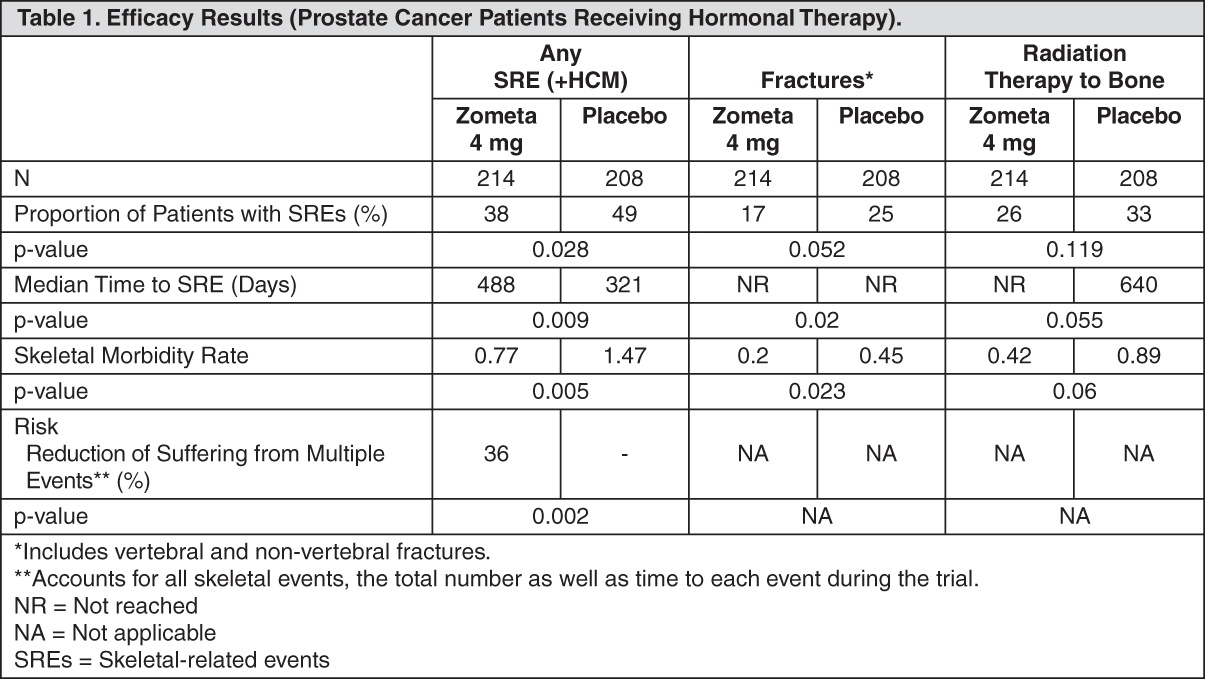 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image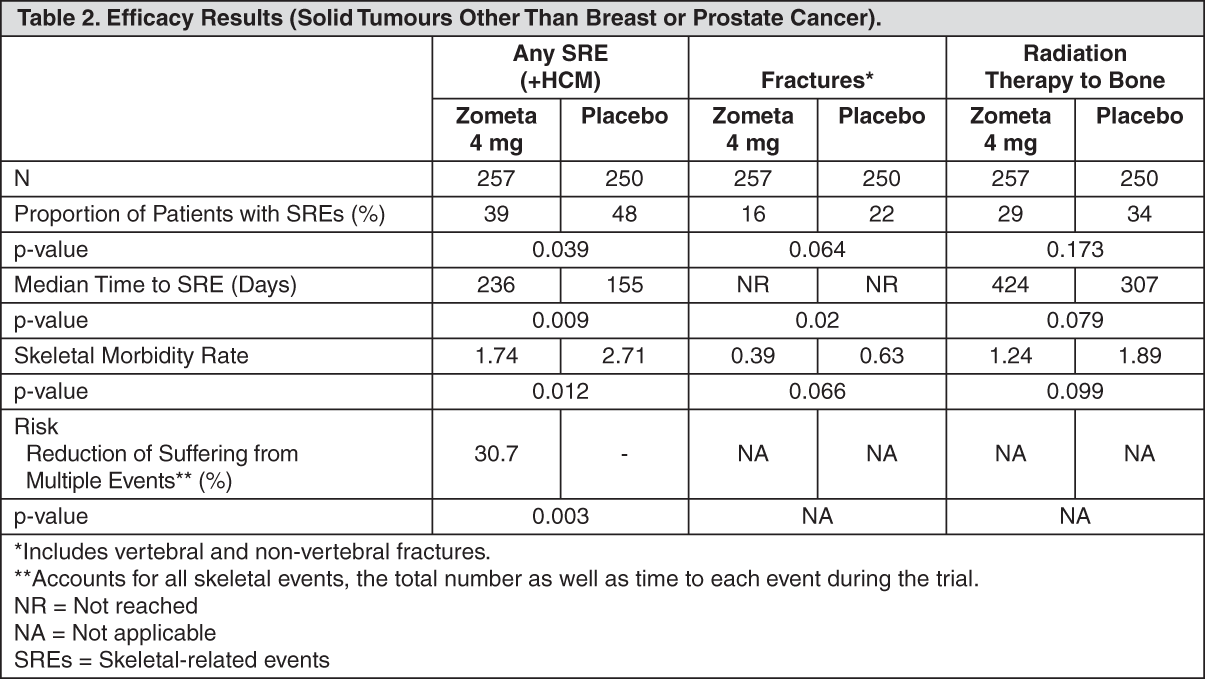 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image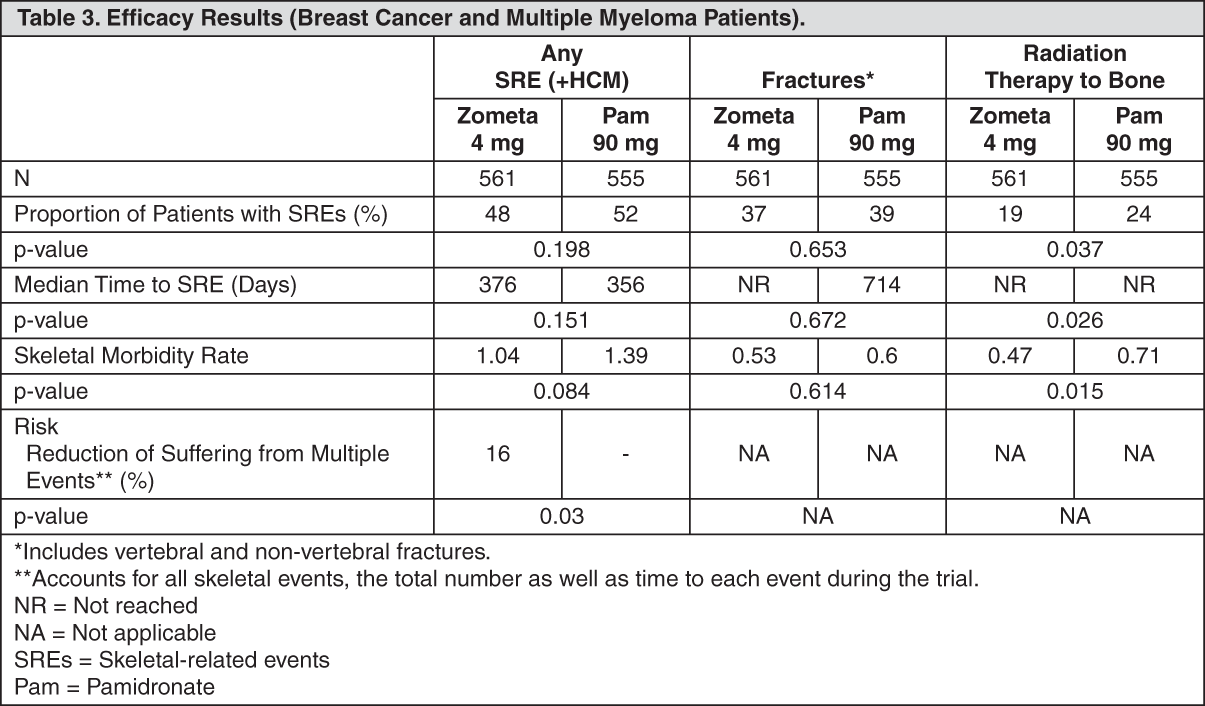 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image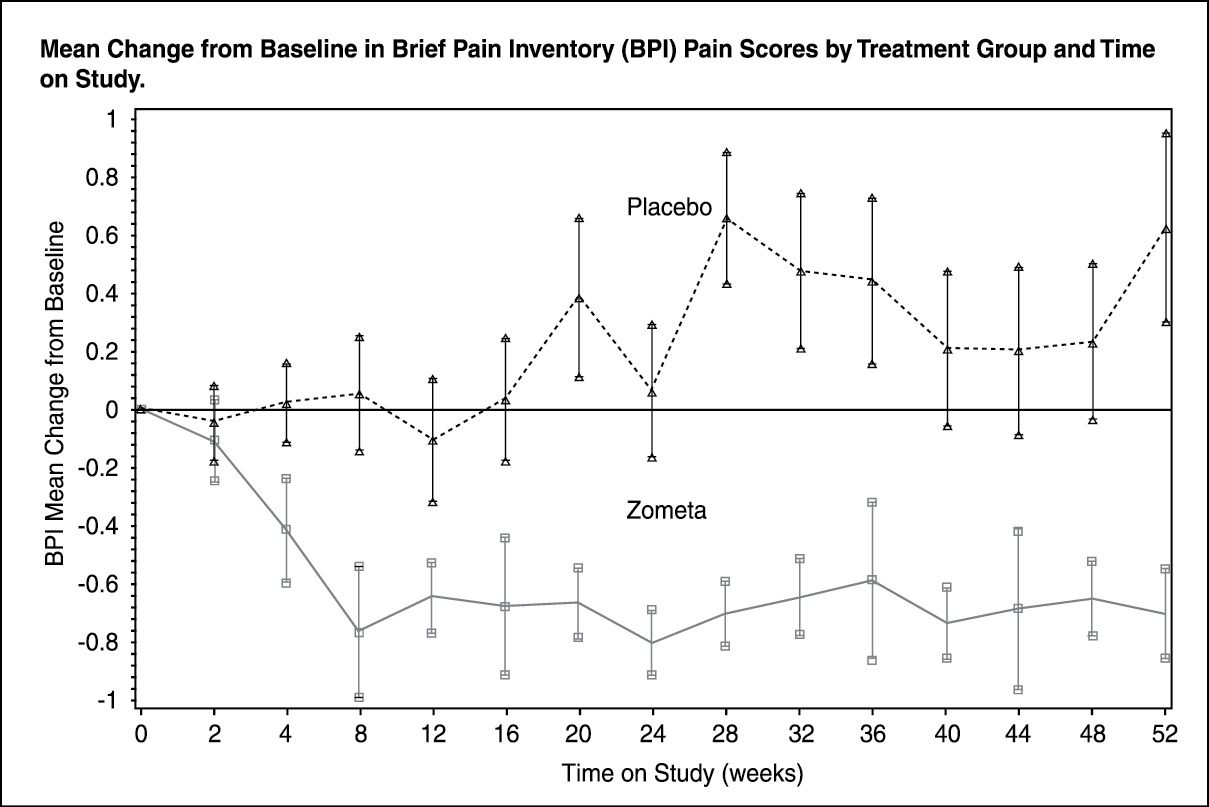 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image


 Sign Out
Sign Out
